




INTRODUCTION
In livestock animals, skeletal muscle is the main component of lean body mass and its growth is the major factor affecting body growth. Generally, the number of muscle fibers is fixed in the prenatal stage, thus postnatal skeletal muscle growth is mainly caused by hypertrophy of existing muscle fibers. Skeletal muscle satellite cells, a kind of adult stem cell, are located between the basal lamina and sarcolemma of the muscle fibers, and have proven to be responsible for skeletal muscle hypertrophy and regeneration (Rhoads et al., 2009). Myogenic satellite cells were first discovered in the frog (Mauro, 1961), followed by numerous distinct methods developed to isolate the muscle satellite cells from different species, e.g. rat (Rosenblatt et al., 1995), human (Blau and Webster, 1981), chicken (Bennett et al., 1986), ovine (Dodson et al., 1986), bovine (Dodson et al., 1987). Porcine skeletal muscle satellite cells were first isolated and cultured in 1992 (Doumit and Merkel, 1992). Afterwards, porcine skeletal muscle satellite cells were isolated using similar or improved methods and were used to investigate the mechanisms of skeletal muscle growth and development (Mesires and Doumit, 2002; Theil et al., 2006; Mau et al., 2008; Wilschut et al., 2010). However, detailed isolation procedures for porcine skeletal muscle satellite cells were not described in these studies, and these methods were time-consuming due to cumbersome steps and produced a low number of satellite cells, which limited the studies on muscle growth and development in pigs. In this study, the isolating processes for porcine skeletal muscle satellite cells were optimized and elaborated in detail, and the characterization of the isolated porcine skeletal muscle satellite cells was further validated. Our study provides a technique for isolation of porcine skeletal muscle satellite cells and will provide material for studying the molecular mechanisms in porcine muscle satellite cells.
MATERIALS AND METHODS
Reagents
Before starting the isolation of porcine muscle satellite cells, the following reagents need to be purchased, including Dulbecco’s Modified Eagle Medium, high glucose (DMEM-HG) (Invitrogen, Grand Island, USA. cat. no. 11995-065), fetal bovine serum (FBS) (Invitrogen, Carlshad, CA, USA. cat. no. 12664-025), horse serum (HS) (Invitrogen, New Zealand, cat. no. 26050-070), 2-[4-(2-hydroxyethyl)-1-piperazine]-1-ethanesulfonic acid buffer (HEPES buffer, 1 M) (Life Technologies, Grand Island, NY, USA. cat. no. 15630106), Phosphate-buffered saline (PBS) (Life Technologies, Grand Island, NY, USA. cat. no. 10010-023), 0.25% Trypsin-ethylenediaminetetraacetic acid (Life Technologies, NY, Grand Island, USA. cat. no. 25200056), Protease from Streptomyces griseus (Sigma, St. Louis, MO, USA. cat. no. P8811, 3.5 U/mg); Collagenase from Clostridium histolyticum (Sigma, St. Louis, MO, USA. cat. no. C7657), 100×Penicillin-Streptomycin (10,000 U/mL) (Invitrogen, Carlsbad, CA, USA. cat. no. 15140-122) and 100% ethanol (GB 678-2002, Beijing, China).
Reagents preparation
Subsequently, the cell culture reagents need to be prepared carefully. The 100×penicillin-streptomycin solution was diluted to 1 fold with PBS to prepare the 1×penicillin-streptomycin PBS (+), and the final concentration of penicillin and streptomycin was 100 U/mL and 100 μg/mL, respectively. The PBS (+) was stored at 4°C until using. The 20% FBS proliferation medium (PM) was prepared with DMEM-HG and FBS according to the ratio of 4:1, e.g. 100 mL 20% FBS PM was prepared with 80 mL DMEM-HG and 20 mL FBS. The PM was stored at 4°C until using or prepared freshly. The 100×penicillin-streptomycin solution was diluted to 1 fold with PM to prepare 20% FBS proliferation medium+ (PM+), the final concentration of penicillin and streptomycin was 100 U/mL and 100 μg/mL, respectively. The PM+ was stored at 4°C until using (not too long before using) or prepared before using. The 2% HS differentiation medium (DM) was prepared with DMEM-HG and HS according to the ratio of 50:1, and the DM was stored at 4°C until using (not too long before using). 1.5 mg proteases (3.5 U/mg) was added to 1 mL preheated PBS including 1.0% HEPES (37°C) to prepare 1.5 mg/mL proteases from streptomyces griseus solution, the solution was gently mixed and filtered through 0.22 μm filter, and stored at −20°C. The collagenase type XI of 1.5 mg was added to 1 mL preheated DMEM-HG containing 5% FBS (37°C) to prepare 1.5 mg/mL collagenase type XI from clostridium histolyticum solution containing 5% FBS, the solution was filtered through 0.22 μm filter, and stored at −20°C. The freezing medium was prepared with dimethyl sulfoxide (DMSO) and FBS according to the ratio of 1:5, e.g. 1 mL freezing medium was prepared with 200 μL DMSO and 800 μL FBS, and then gently mixed. The freezing medium was prepared before using. The ethanol of 75% and 70% was prepared with 100% ethanol and sterilized ultrapure water according to the ratio 3:1 and 7:3, respectively. E.g. 100 mL 75% ethanol was prepared with 75 mL 100% ethanol and 25 mL sterilized ultrapure water, and the solution was gently mixed.
Instruments and consumables
The following instruments, including water bath, centrifuge, incubator, light microscope, confocal microscopy, ice maker, are required for the isolation of porcine muscle satellite cells. In addition, the following consumables also need to be prepared, including scalpels, razor blades, forceps, scissors, cell strainers (pore size 40 μm, 70 μm, 100 μm), plastic petri dishes, polypropylene centrifuge tubes (15 mL, 50 mL), culture flask (T75, T25), Cryo tube vials, 6-wells culture dishes. All the consumables must be sterile and then can be directly used for cell culture.
Pig muscle tissues collection
The Duroc×Yorkshire×Landrace newborn piglets less than one week old were provided by Jiangpu Farm of Nanjing Agricultural University. The piglets were slaughtered at College of Animal Science and Technology of Nanjing Agricultural University according to the Animal Care and Use Statute of China, and all the sample collections were specifically approved by the ethics committee of Nanjing Agricultural University.
After slaughter, the body surface of the piglets was sterilized with 75% ethanol, and then transferred in to the cell culture laboratory for muscle tissue collection. The hair on hind legs was removed and the skin of the hind legs sterilized with 75% ethanol, then the whole semitendinosus (ST) and semimembranosus (SM) muscles on the right and left were dissected using scalpels, respectively.
Isolation and culture procedures of muscle satellite cells
Firstly, the collected muscles were quickly sterilized with 70% ethanol in a plastic petri dish, and then immediately transferred into a new plastic petri dish with cold PBS (+) and rinsed 3 to 4 times with 4 fold volume cold PBS (+) in a new plastic petri dish. Visible adipose and connective tissues on the muscle mass were removed with a scalpel in laminar-flow hoods or Biological safety cabinets.
Secondly, the whole ST or SM muscles were excised and cut into small pieces using scissors under the cold PBS (+) containing 1% HEPEs in a plastic petri dish and the shredded muscle pieces were transferred into 15 mL polypropylene centrifuge tubes, centrifuged at 1,000 g for 5 min to separate the supernatant and shredded muscle pellet. The supernatant was again centrifuged at 2,000 g for 5 min to collect a second pellet. Then 2 to 4 folds volume 1.5 mg/mL Protease solution was added to the pooled collected pellets (ensuring the muscle pieces were covered by protease solution).
Thirdly, the 15 mL polypropylene centrifuge tubes containing the muscle pieces were digested in a water bath at 37°C for 1 h with 10 min shaking (a continuous shaking water bath would be better) and the digested muscle pieces were aspirated back and forth for 10 to 15 times with 5 or 25 mL pipettes to gently dissociate the cells, and centrifuged for 5 min at 200 g to separate the supernatant containing the cell pellet and the underlying tissue fragments pellet. While the underlying tissue fragment pellets were temporarily kept on ice for subsequent collagenase type XI digestion. Then, the supernatant was transferred into 15 mL polypropylene centrifuge tubes and was centrifuged for 5 min at 2,000 g to collect the cell pellet, and the cell pellet was further resuspended with 10 mL PM+, and then centrifuged for 10 min at 2,000 g to collect the cell pellet again. Afterwards, the cell pellet was resuspended again with 5 mL PM+, and the cell suspension was filtered through 100 μm and 70 μm cell strainers, successively. Then, the filtered cell suspension was centrifuged for 10 min at 2,000 g to collect the cell pellet (Pellet I), and the cell pellet was resuspended with 1 mL PM+ again and kept on ice.
Fourthly, the underlying tissue fragment pellet derived from the third step was further digested for 1 h with 2 to 4 folds pre-prepared collagenase type XI solution. Similarly, the tissue fragments were aspirated for 10 to 15 times with 5 or 25 mL pipettes to gently dissociate the cells and filtered through a 100 μm cell strainer, Then, the filtrated cell suspension was centrifuged for 5 min at 2,000 g to separate the supernatant and the cell pellet, and the supernatant centrifuged for 10 min at 2,000 g to collect the residual cell pellet, and both collected cell pellets were mixed. Next, the cell pellet was resuspended with 10 mL PM+ in 15 mL polypropylene centrifuge tubes and the cell suspension was filtered through a 70 μm cell strainer, and then centrifuged for 5 min at 2,000 g to collect the cell pellet (Pellet II). The cell pellet was resuspended with 1 ml PM+ and kept on ice.
Fifthly, the cell suspension (Pellet I and Pellet II) were mixed in 15 mL polypropylene centrifuge tubes and centrifuged for 5 min at 2,000 g to recover the Pellet I and Pellet II. Next, the cell pellet was resuspended with 10 mL cold PBS (+) and centrifuged for 10 min at 2,000 g to collect the cell pellet. Subsequently, the cell pellet was resuspended with 5 mL PM+ again and the cell suspension was filtered through a 40 μm cell strainer. Then, the cell suspension was adjusted to 10 mL with PM+, centrifuged for 10 min at 700 g to collect the cell pellet.
Finally, the cell pellet was resuspended in 10 mL 37°C preheated PM+ and the cell suspension was pre-plated in T75 cell culture flask and incubated at 37°C under 5% CO2 for 1 h. The fibroblasts become quickly adherent to the bottom of cell culture flask, while the skeletal muscle satellite cells will remain in the supernatant. Then, the supernatant containing the skeletal muscle satellite cells was collected to 15 mL polypropylene centrifuge tube and centrifuged for 10 min at 500 g, and the cell pellet was resuspended with 10 mL PBS+, centrifuged for 10 min at 500 g to collect the skeletal muscle satellite cells pellet. Afterwards, the skeletal muscle satellite cells pellet was resuspeneded with 10 mL 37°C preheated PM+ and plated in T75 cell culture flask and incubated at 37°C under 5% CO2.
Identification of muscle satellite cells
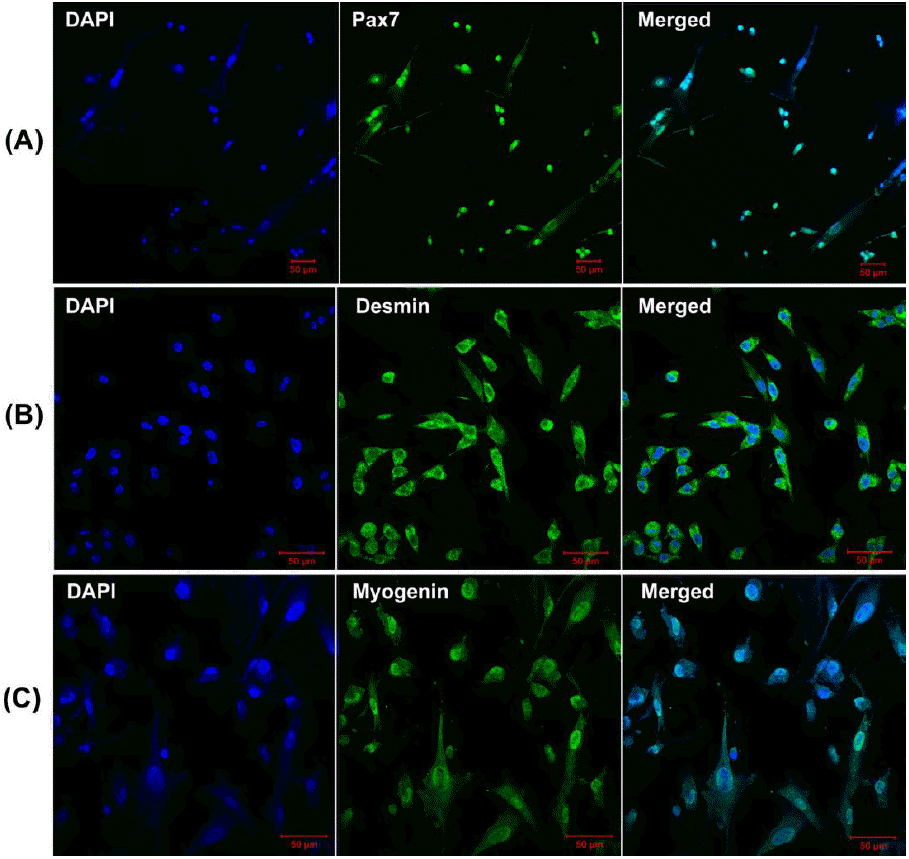
To characterize the porcine skeletal muscle satellite cells, the isolated cells were plated into covered 6-well plates at a density of 105 cells/mL per well and cultured in an incubator at 37°C with 5% CO2 until to 70% to 80% confluent. Subsequently, immunofluorescence staining with specific antibodies was performed to identify the porcine skeletal muscle satellite cells. Briefly, the cells were washed three times with PBS and fixed with 4% paraformaldehyde for 30 min at room temperature, and then washed three times for 5 min each with PBS again, followed by permeabilization with 0.5% Triton X-100 for 30 min at room temperature. Afterward, the cells were blocked with 1% bovine serum albumin (BSA)-supplemented PBS for 1 h and incubated overnight at 4°C or 4 h at room temperature with anti-Pax7 primary antibody (Santa Cruz Tech, Santa Cruz, CA, USA) and anti-Desmin primary antibody (Santa Cruz Tech, USA). After three washes for 5 min each time in PBS, the cells were labeled with appropriate fluorescein-labeled secondary antibody for 1 h in the dark at room temperature, and followed by washing three times in PBS again (5 min each time). Meanwhile, the cell nuclei were counterstained with 4′, 6-diamidino-2-phenylindole for 5 min, and then the samples were mounted on glass slides and examined on confocal laser scanning microscope (Zeiss LSM 700 META, Jena, Germany).
Proliferative properties of muscle satellite cells
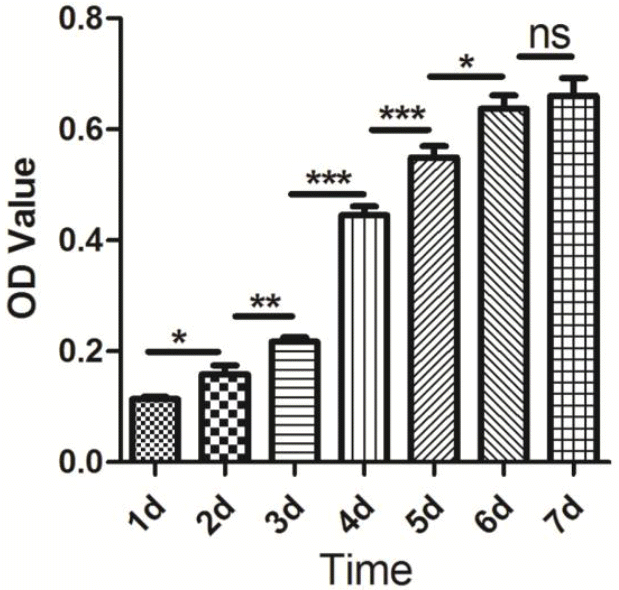
Proliferative properties of the porcine skeletal muscle satellite cells were detected using cell counting kit-8 (CCK-8) (Dojindo, Tokyo, Japan). The isolated cells were seeded into 96-well plates at a density of 2×103 cells/well with 100 μL culture medium. After 1, 2, 3, 4, 5, 6, and 7 d of incubation at 37°C with 5% CO2, respectively, 10 μL CCK-8 solution was added to each well and incubated in 5% CO2 at 37°C for 4 h. Finally, absorbance was measured at 450 nm using a Multiskan GO Microplate Spectrophotometer (Thermo Scientific, Waltham, MA, USA). All tests were carried out in ten replicates and the data were presented as means±standard error. All statistical analyses were performed using SPSS version 18.0. Differences between two adjacent groups were evaluated using unpaired-sample t-test. A p value <0.05 is shown as *, p<0.01 is shown as **, and p<0.001 is shown as ***.
Differentiation characteristics of muscle satellite cells
To validate the differentiation characteristics of skeletal muscle satellite cells, the isolated cells were seeded into the 6-well plates at a density of 104 cells/mL per well and were cultured with DMEM supplemented with 10% FBS in an incubator at 37°C with 5% CO2 until 50% confluent. Then, the cells were cultured with DMEM containing 2% HS to induce differentiation and the cell morphology was observed with an inverted microscope (Leica fluorescence microscope, Mannheim, Germany). Moreover, the differentiation characteristics of muscle satellite cells were confirmed with differentiation biomarker protein anti-myogenin (Santa Cruz, USA) using immunofluorescence staining. The protocol was performed according to the method mentioned above.
RESULTS AND DISCUSSION
Skeletal muscle satellite cells are a useful model to investigate the mechanisms of muscle growth and development. Pigs are one of the most important economic animals and make a substantial contribution towards meeting human nutritional requirements. Therefore, the study of porcine skeletal muscle growth and development using the porcine skeletal muscle satellite cell model is of great importance.
The earliest skeletal muscle satellite was found in an electron microscopic study of the peripheral region of the frog skeletal muscle fiber (Mauro, 1961), but the first viable satellite cells were isolated from adult rat skeletal muscles (Bischoff, 1974). Afterward skeletal muscle satellite cells from different species were isolated according to the protocol established by Bischoff in 1974, while the isolation and culture of porcine skeletal muscle satellite commenced in 1992 (Doumit and Merkel, 1992). Although the isolating and culturing protocols for skeletal muscle satellite cells were established in many species, the processing conditions may differ considerably between species. Furthermore, methods have been updated continuously with the developments inbiotechnology. Therefore, updates of these methods will accelerate the research progress of skeletal muscle growth and development using satellite cells model. At present, two major approaches have been applied to isolate skeletal muscle satellite cells, the first approach is to break down the connective tissue network and myofibers to release the muscle satellite cells based on the mincing, enzymatic digestion and repetitive trituration of the muscle mass. This is the classical and efficient method to obtain enough muscle satellite cells, although this method may obtain a heterogeneous population of precursor cells. The second approach is to isolate the muscle satellite cells from single intact muscle fiber, which can obtain relatively pure muscle satellite cells, and this method has been successfully used in studies of muscle satellite cells in rats (Kastner et al., 2000), mice (Shefer and Yablonka-Reuveni, 2005) and humans (Bonavaud et al., 2002). In the present study, the approach was based on enzymatic digestion to isolate the porcine skeletal muscle satellite cells. In the isolating process, proteases from Streptomyces griseus and collagenase type XI were chosen to digest the skeletal muscle tissues. The concentration and digestion time were optimized to achieve the satisfactory dissociation and viability cells according to the characteristics of muscle tissues and the product manual.
Skeletal muscle satellite cells are a kind of adult stem cells, therefore postnatal stages were suitable for the isolation of skeletal muscle satellite cells. The previous studies indicated that the absolute number increased between 1 and 32 weeks of age. This number was at least maintained between 1 and 64 weeks of age; however, the relative proportion of porcine skeletal muscle satellite cells gradually decreased from 1 to 64 weeks after birth (Campion et al., 1981; Mesires and Doumit, 2002). Therefore it is better to select newborn piglets at no more than two weeks of age to obtain a high proportion of muscle satellite cells. In this study, one week old piglets were used to isolate the skeletal muscle satellite cells and good positive skeletal muscle satellite cells were obtained.
Skeletal muscle satellite cells are located between the basal lamina and sarcolemma of the muscle fibers. While the number of muscle satellite cells may differ considerably among specific skeletal muscles in different parts of body, e.g. the extensor digitorum longus (EDL) and the soleus (SOL) muscle in mouse, the number of satellite cells in SOL is greater than in EDL from 2 to 3 months old mice (Di Foggia and Robson, 2012). Notably, the hindlimb muscles are generally used to isolate the skeletal muscle satellite. For example, the SM and ST muscles are mostly used for the isolation of skeletal muscle satellite cells in pigs (Doumit and Merkel, 1992; Blanton et al., 1999; Mesires and Doumit, 2002; Mau, Oksbjerg et al., 2008; Wilschut et al., 2008), while the limb muscles extensor EDL, tibialis anterior, and flexor digitorum brevis are used to isolate the skeletal muscle satellite cells from mice (Shefer and Yablonka-Reuveni, 2005). In the present study, porcine SM, ST and longissimus dorsi muscles were chosen to isolate muscle satellite cells. Our results indicate that the muscle satellite cells can be isolated from three kinds of muscles, although we did not provide the evidence indicate which one is better. The phenotype of proliferating and differentiated porcine skeletal muscle satellite cells were shown in Figure 1.
At present, although several proteins have been demonstrated to characterize skeletal muscle satellite cells, including Pax7, M-cadherin, Cxcr4, syndecan3/4, and c-met (Fukada et al., 2013), Pax7 is the most commonly used molecular marker used to characterize the skeletal muscle satellite cells derived from distinct species. In our study, Pax7 and Desmin antibodies were used to characterize the proliferating positive muscle satellite cells, while myogenin antibody was used to monitor the differentiation capability of muscle satellite cells using immunofluorescence. The characterized results from immunofluorescence were shown in Figure 2. Furthermore, the result of CCK-8 analysis indicated that the isolated skeletal muscle satellite cells display normal proliferative capacity (Figure 3). Taken together, these results indicate that the optimized method can obtain high pure skeletal muscle satellite cells which can be utilized as cell model to investigate the molecular mechanisms of skeletal muscle growth and development.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







