





INTRODUCTION
Meat quality in chickens is an important trait and includes pH, meat color, drip loss, tenderness, intramuscular fat content, and other fat traits such as the contents and proportions of abdominal and subcutaneous fat. Modern broilers grow very fast due to genetic selection, efficient production systems, improved nutrition and regular veterinary attention. However, selection for fast growth and high yield may have negatively impacted qualities of the meat [1]. The elucidation of the molecular mechanisms underlying meat quality traits in chickens will have both biological and economic consequences.
Previously, quantitative trait loci (QTLs) have been studied for many traits in chicken for over 20 years to identify genetic markers and functional factors for physiological mechanisms. A variety of QTLs were detected for meat quality traits and abdominal fat traits throughout various genomic regions [2]. These QTLs were detected by linkage analysis and by candidate gene analysis. Both of these methods have limitations: the identified QTL regions are generally large and require subsequent fine mapping to identify closely linked markers or causative variants. Candidate genes, based on putative physiological roles, may exclude the identification of novel genes or pathways that influence the target traits [3]. Recently, genome-wide association studies (GWAS) in an F2 broiler resource population identified 14 new genes for several meat quality traits [4]. Most of the regions identified by GWAS were found in regulatory regions which make the identification of causal mutations (e.g. coding sequence changes, deletions, or duplications) difficult.
Two broiler chicken lines divergently selected for muscle color (high muscle color [HMC] and low muscle color [LMC]) along with their random bred control line (RAN) have been developed at University of Arkansas, Fayetteville, Arkansas, United States [4,5]. HMC and LMC lines have undergone 8 generations of selective pressure based on muscle color and those lines clearly showed differential muscle qualities [5]. After 8 generations of divergent selection for muscle color, HMC lines showed ~4.2 higher L* value compared to parental RAN line while LMC lines showed ~2.8 lower L* values compared to RAN line [5]. When compared, the L* value for the HMC line (53.91┬▒0.28) was ~7.1 higher than the LMC line (46.86┬▒0.20). This result clearly showed that the meat color is different between HMC and LMC lines.
In this study, whole genome re-sequencing analysis using Illumina sequencing platform was performed to investigate genetic variation for muscle color expression in HMC and LMC lines compared with their parental random bred line, RAN. Millions of single nucleotide polymorphisms (SNPs) were identified by genome re-sequencing and only potentially causal genes containing non-synonymous mutations, which can induce amino acid changes in proteins were detailed in this study.
MATERIALS AND METHODS
Genetic lines and Illumina sequencing
HMC and LMC lines were formed from the divergent selection of muscle quality on a random bred control line (RAN) that has been maintained by N. B. Anthony at the University of Arkansas. L* value is a key visual indicator to determine muscle color of breast fillet using Minolta CR-300 Colorimeter (Minolta Italia S.P.A, Milano, Italy) and selection methods and line development were reported previously by Harford et al [5]. Bird populations in the 8th generation of selection were used for genome sequencing. Blood (3 mL) was collected from 12 birds each following animal use protocol approved by IACUC (University of Arkansas; Project number: 11025). Genomic DNA was isolated from each whole blood sample using QiaAmp DNA mini kit (Qiagen, Valencia, CA, USA) following manufacturerŌĆÖs instructions. DNA quality was determined by agarose gel electrophoresis and 10 samples having the highest qualities in each line were pooled to represent their respective line. Library preparation and Illumina genome sequencing for the pooled DNA samples were performed by the National Center for Genome Resources (NCGR; Santa Fe, NM, USA). Illumina HiSeq system 2├Ś100 bp paired end read technology was used for genome sequencing.
Genome sequence assembly and data analysis
Quality of raw reads were determined using FastQC tool kit (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and adapters were trimmed out using bbduk.sh command line of bbmap toolkit (https://sourceforge.net/projects/bbmap/). Cleaned reads were aligned to the galgal4 chicken reference genome sequence that was retrieved from National Center for Biotechnology Information. For the reference based genome alignment, the NGen genome sequence assembly program of the Lasergene software package (DNAStar, Madison, WI, USA) was used. Assembly parameters were as follows: file format, BAM; mer size, 21; mer skip query, 2; minimum match percentage, 93; maximum gap size, 6; minimum aligned length, 35; match score, 10; mismatch penalty, 20; gap penalty, 30; SNP calculation method, diploid bayesian; minimum SNP percentage, 5; SNP confidence threshold, 10; minimum SNP count, 1; minimum base quality score, 5. After assembly, the SeqMan Pro program of the Lasergene package was used for further analyses including SNP data.
SNP detection and analysis
SNP calling and further analysis were following previous report [6]. Briefly, JMP Genomics (SAS Institute Inc., Cary, NC, USA) program was used for filtering unique SNPs for HMC and LMC chickens. SNPs occurring in both HMC and LMC lines were filtered out, leaving behind unique SNPs for each line with relatively high SNP quality call (Q call >40 out of highest value = 60). To call highly fixed and homozygous SNPs, SNP percentages (SNP%) was the next filtering point, anything less than 75% (for example, number of SNP = 3 of read depth = 4) was not considered. The cutoff of Q call >40 and SNP% >75% for SNP selection was set by considering potential sequencing errors that can be generated by the massively parallel sequencing method. Then, chicken line specific SNPs were determined for the segregation of parental- (RAN) or non-parental mutations. To focus on the novel mutations that are different from parental genotypes, non-parental SNPs were chosen in each line. Potential causal SNPs showing non-synonymous changes in coding DNA sequences (CDS) region were chosen in each line. Since read depth of many SNPs were low range, unique SNPs showing over 10 depths were considered as reliable SNPs specific for each genetically selected line. Reliable, mutational and causal SNPs, which were chosen by criteria described above were confirmed by double-checking the raw assembly data with alignment view to reduce false positives.
Bioinformatic analyses
The effect of SNP variants (non-overlapping, >40 Q call, >75 SNP%, >10 depths and non-synonymous changes) in protein functionality was determined using Ensembl Variant Effect Predictor (http://useast.ensembl.org/info/docs/tools/vep/index.html). Functional interpretation of genes retaining SNPs was analyzed in the context of gene ontology and molecular networks using the ingenuity pathways analysis (IPA; Qiagen; www.ingenity.com). Since IPA is based on human and mouse bioinformatics, functionalities for selected genes in the chicken were interpreted based primarily on mammalian biological mechanisms. The number of molecules in the network was set to the limit of 35, leaving only the most important ones based on the number of connections for each focus gene (a subset of uploaded significant genes having direct interactions with other genes in the database) to other significant genes [7].
RESULTS AND DISCUSSION
Genome re-sequencing for HMC, LMC, and parental RAN line chickens
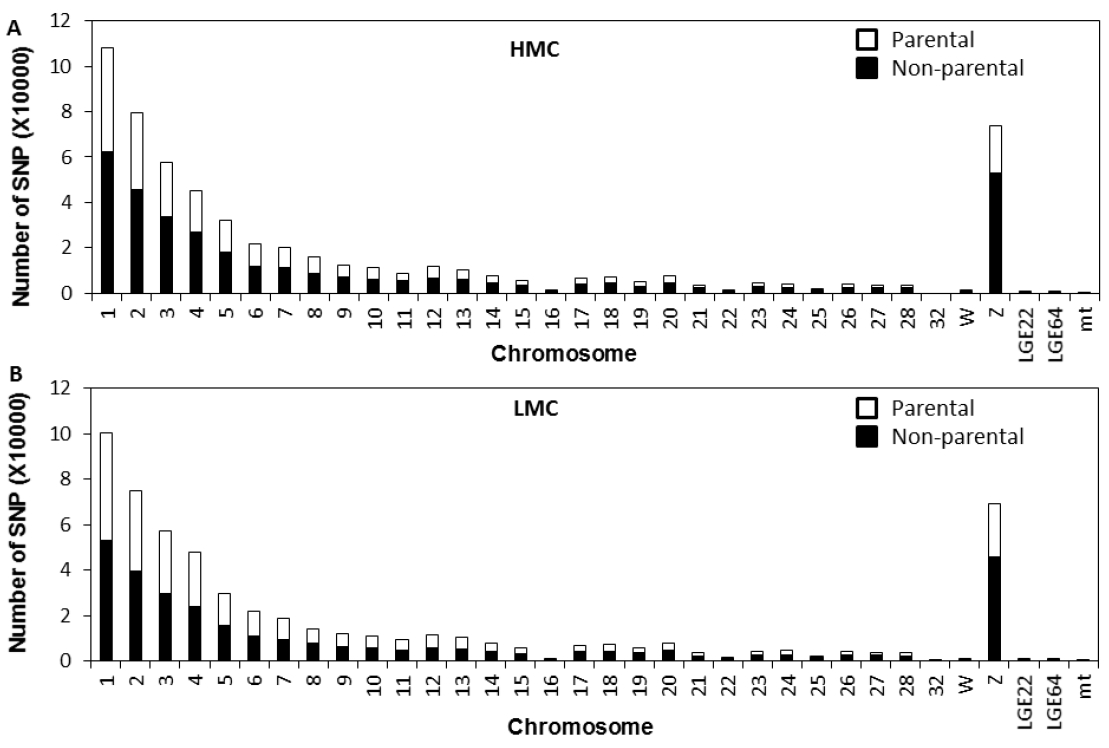
Results of genome sequencing of pooled DNA from each of 10 chickens per line showed 5.1├Ś, 6.5├Ś, and 8.2├Ś genome coverage for HMC, LMC, and RAN lines, respectively. The total number of SNPs was 5.3, 6.0, and 4.4 million (~0.5% of template genome) in HMC, LMC, and RAN genome, respectively (data not shown). The large number of SNPs per examined chicken line was based on data of at least 2 read coverage depths (number of read counts per nucleotide location). To identify genetic biomarkers that are responsible for regulating muscle color, high quality (Q call >40) SNPs uniquely found in HMC and LMC were selected by removing overlapping SNPs between HMC and LMC simultaneously. Then, possibly fixed mutations showing >75% SNP rates were chosen as reliable marker SNPs. As a result, a total of 1,134,655 unique SNPs were identified throughout the HMC and LMC chicken genomes. The number of SNPs in each chromosome is shown in Figure 1. The HMC line (Figure 1A) showed 576,886 SNPs including 229,415 parental and 347,471 non-parental segregations, while the LMC line (Figure 1B) showed 557,769 SNPs including 253,055 parental- and 304,714 non-parental segregations (Information for all SNPs for HMC and LMC will be provided upon request). When SNPs were grouped by feature types of chromosome regions, ~50% of SNPs were in the intergenic (heterochromatic) regions and 26,800 SNPs were found in CDS (protein coding regions). Around 70% of SNPs in CDS regions were synonymous mutations that do not induce amino acid changes. A total of 2,884 SNPs (1,307 for HMC and 1,577 for LMC) could induce non-synonymous-, frameshift-, nonsense-, and no-start mutations in proteins, suggesting that the 2,884 SNPs may be inducible mutations that are part of the genetic components regulating muscle color in chickens. Of those 2,884 candidate SNPs associated with amino acid changes, 679 and 924 SNPs were parental (RAN line) segregations for HMC and LMC, respectively (data not shown; information for all SNPs for HMC and LMC will be provided upon request). To focus on the novel mutations different from parental genotypes, SNPs of non-parental segregation showing non-synonymous changes in CDS region were considered as causal mutations in this study. Potentially causal SNPs showing over 10 read depths (considered to be more reliable candidate genetic markers) were chosen for further bioinformatic analysis as described in Materials and Methods. In addition, re-scanning of each SNP position for reliable, mutational, and causal protein coding SNPs was conducted to reduce false positives due to possible errors (e.g. SNP detection by reading a position in a chicken line, which was not covered the region of the SNP in the other chicken line) in the SNP calling process, using Seqman-Pro viewer program. This process yielded 15 more reliable SNPs including 1 for HMC and 14 for LMC (Table 1) and gene names containing SNPs that are listed in Table 2. Genes included are: trafficking protein particle complex 8 (TRAPPC8) for HMC and apolipoprotein B antigen (APOB), ataxia telangiectasia mutated (ATM), coiled-coil domain containing 88A (CCDC88A; transcript variant 1), complement factor H (CFH), collagen, type XXVIII, alpha 1 (COL28A1), glycoprotein IX (GP9 [platelet]), ligase IV (LIG4, DNA, ATP-dependent), melanoma inhibitory activity family, member 3 (MIA3), microtubule associated monooxygenase, calponin and LIM domain containing 2 (MICAL2), RE1-silencing transcription factor (REST), small nuclear RNA activating complex, polypeptide 1 (SNAPC1, 43 kDa; transcript variant 1), TNF receptor-associated factor 1 (TRAF1), UTP18 small subunit processome component homolog (UTP18 [yeast]), and LOC428119 (uncharacterized protein) for LMC.
The effect of SNP variants in the protein functionality of genes listed above was determined using Ensembl Variant Effect Predictor, resulting in that two SNPs found in ATM and APOB can induce deleterious amino acid changes, while others were tolerated mutations (data not shown). Both mutations in ATM and APOB were found in armadillo (ARM) repeats superfamily domain (superfamily ID: SSF48371 given from http://supfam.org/SUPERFAMILY/index.html).
Bioinformatic analyses with genes retaining SNPs associated with muscle color
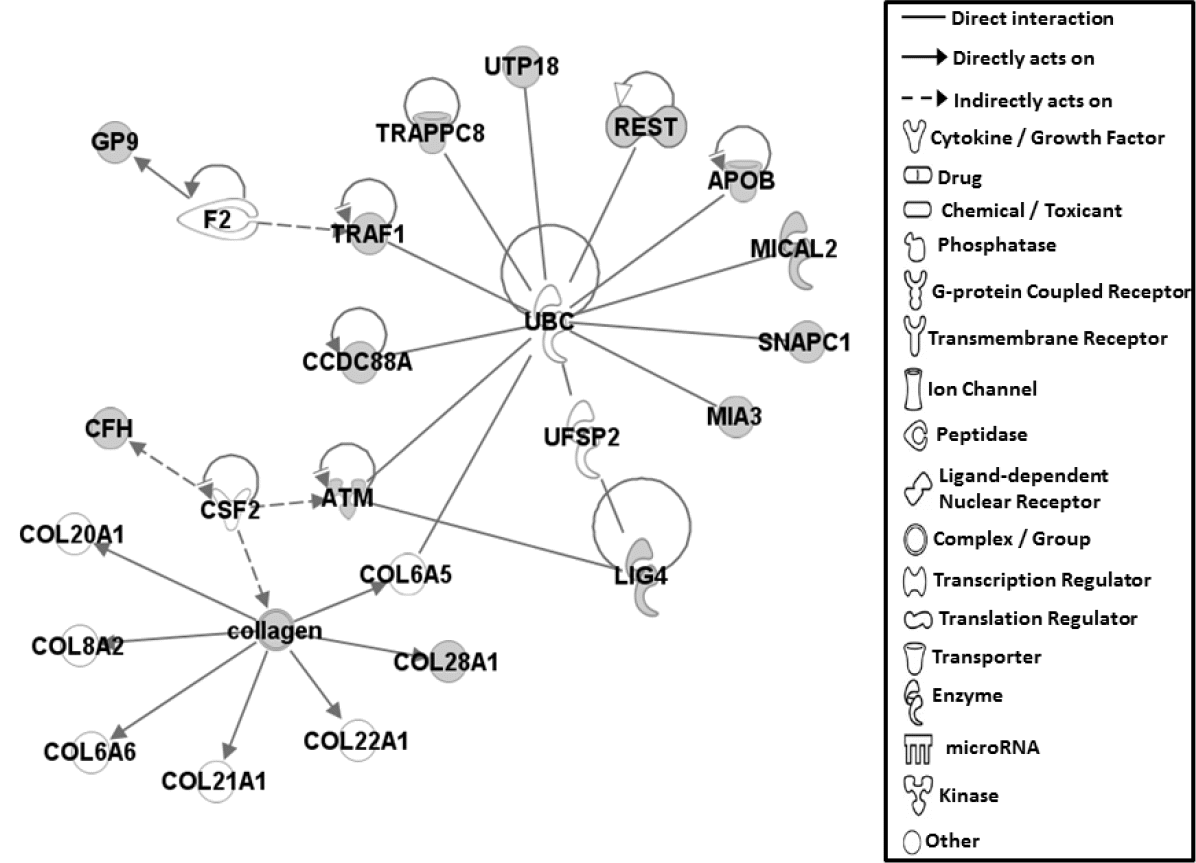
Fifteen genes containing potentially causal SNPs, listed above, were subjected to gene network analysis, which represents the intermolecular connections among interacting genes based on functional knowledge inputs, using the IPA program. Of various assay settings, the simplest settings of 35 focus molecules were employed to analyze molecular gene networks that resulted in only one network being generated (Figure 2). ATM and LIG4 are associated with cellular assembly and organization. ATM, a protein kinase family member plays a critical role in the cellular response to DNA damage by phosphorylating downstream targets such as BRCA1 (breast cancer 1, early onset) and p53 [8], in addition to a direct role in telomere maintenance [9]. LIG4 mainly functions in repair of DNA double-strand breaks by non-homologous end joining process in the maintenance of genome stability [10]. Moreover, CCDC88A, also known as Akt-phosphorylation enhancer (APE) can decreases DNA synthesis with suppression of protein kinase B phosphorylation [11]. Taken together, potentially causal amino acid mutations in molecules involved in DNA synthesis, telomere maintenance and DNA damage responses appear to be associated with muscle color expression in broiler chickens. However, the direct connection of phenotypic expression of muscle color to basic cellular function for chromosomal DNA integrity, cellular structure, and proliferation is still unclear and further investigations are needed to discover genetic markers and to understand molecular regulation of muscle color expression. Molecules, such as APOB, ATM, MIA3, MICAL2, REST, TRAF1, UTP18, TRAPPC8, CCDC88A, and SNAPC1 in the network were shown to bind directly to UBC (ubiquitin C) by proteomic analyses of ubiquitylated proteomes [12ŌĆō15], suggesting that the various cellular functions including protein degradation by ubiquitylation may play a role for the expression of muscle color in chickens though the specific roles of each factor in regulating muscle color development need further investigations. In addition, membrane bound- and secreting proteins, such as APOB, CFH, GP9, and COL28A1 were shown to be associated with regulating muscle color. Secreting proteins can be used as biomarkers in the biofluid for muscle color trait. Specifically, GP9 is found in the platelets of blood, thus amino acid difference in GP9 can be used as blood biomarker to select muscle color traits in chickens [16]. Previous results of GWAS for meat quality in chicken identified 14 marker genes including a collagen gene (collagen, type 1, alpha 2) [4]. The extracellular matrix of muscle is composed mostly of the protein collagen family and muscle qualities can be reflected by the relative amount and distribution of collagen fibers in muscles depending on genetic, physiological and nutritional conditions [17]. This suggests that genetic alteration in the collagen gene and the contents of collagen proteins may function in regulating meat quality, especially meat color.
It is hypothesized that the SNPs found in this study are the direct result of divergent selection for L*, however, one cannot ignore the potential impact of random genetic drift. Each research line has been maintained with 24 sires and 3 dams per sire providing an effective population size of 72. This is sufficient to manage inbreeding and significant impacts of drift in research populations [18]. It is possible, however, that sampling error could have been introduced by the initial pooling of 10 samples per line. One could further validate our results by testing a larger number of birds using potential marker candidates, that are identified by the genome-wide screening methods.
In this study, various potential genetic markers showing amino acid changes were identified in differential meat color lines, HMC and LMC, through genome re-sequencing. When considering the functional standpoint based on the interpretation of factors involved, meat color in chickens appears to be associated with chromosomal DNA stability, functions of ubiquitylation (UBC) and quality and quantity of various subtypes of collagen in muscle. Since wide-spread SNPs throughout genomic regions can influence the expression of muscle color by various mechanisms, such as altered gene expressions (e.g. SNPs in promoter regions may regulate expressions of mRNAs and proteins), the genes listed in this study may not cover all potentially causal mutations in muscle color change. However, non-synonymous mutations induced by SNPs in protein coding regions, that were mainly focused in this study, may become critical determinants in protein structures and their functionalities for the expression of muscle color. In this regard, the SNPs causing amino acid changes were analyzed first in this study and further studies will be conducted to characterize SNPs found in other feature regions of genome. Additionally, functional validation studies for the candidate factors will follow, using the muscle color chicken lines.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print







