




INTRODUCTION
The pig gastrointestinal tract harbors thousands of species of bacteria, whose composition and relative proportions vary depending on the animal breed, animal age, and nutritional and environmental factors [1]. Gut bacteria influence nutrient absorption and the health of the host [2,3], such as vitamin synthesis and short-chain fatty acid (SCFA) production [4]. In particular, genetic background is strongly associated with the hostŌĆÖs gut microbial taxa and characteristics [2,4].
Hampshire, Landrace, Duroc, and Yorkshire are the most frequently used pigs in commercial production and have favorable growth performance. These breeds had various origins. Duroc pigs are red, muscular pigs that originated in America. Yorkshire, also known as Large White, is a breed originating in Yorkshire. The Hampshire pig has a black body with a white band covering the front legs and originated from America. The Landrace breed was developed in Denmark by crossing the native pig with the Large White and has white skin. Landrace has bigger size and faster growing rate but low ability of resistance to disease [5]. Overall, male pigs utilize feed more efficiently and have profitable production characteristics, but few studies have compared the fecal bacteria compositions of these four boar breeds [6]. In this study, the four purebred boar pig lines were housed in a controlled environment and fed the same diet. Due to their distinct genetic backgrounds, the four breeds of pigs exhibit differences in morphology, reproduction and digestive capacity. Therefore, these breeds of pigs may exhibit breed-specific microbial diversity [2,7]. The Illumina Sequencing platform yields longer deoxyribonucleic acid (DNA) reads at lower cost compared to other platforms and provides more abundant information on bacterial communities [3]. However, given the diverse relationships between boar pig properties and gut microbiome function, further study is needed to reveal the contribution of gut microbiome and its metabolic products to an animalŌĆÖs economic characteristics.
To assess the influence of pig breed on shaping the gut micro biota and to determine whether these breeds share core bacteria or have breed-specific bacteria, we chose twenty-three boar pigs (Duroc = 6, Hampshire = 4, Landrace = 7, Yorkshire = 6) representing four breeds and diverse genetic backgrounds. With the help of sequence statistics, we identified ten predominant genera that co-occurred in all pig breeds and several breed-specific bacteria that may be potentially functional microorganism to pig breeds.
MATERIALS AND METHODS
Sample collection and DNA extraction
Fresh fecal samples were collected from four purebred boar pigs approximately 300 days of age. Pigs were raised by a method approved by the Animal Care and Use Committee of ZAAS (Approval number: ZAAS-2015-008). The four breeds were raised in under standard American facility farming (Tonglu, Zhejiang, China), and all pigs received the same primary diet based on corn and soybean without antibiotics. All pigs were grown in the same hogpen. The temperature and humidity of the house were controlled at comfortable levels (temperature was 16┬░C to 21┬░C and humidity was 60% to 80%). All samples were collected on the same day in the morning before feeding and frozen in liquid nitrogen before immediate transport to the laboratory. Total genomic DNA was extracted from the samples using the QIAamp DNA Stool Mini Kit (QIAGEN, CA, Hamburg, Germany) according to the manufacturerŌĆÖs instructions [2]. The DNA concentration and purity were monitored on 1% agarose gels. The quantity of DNA was measured using a NanoDrop 1000 spectrophotometer, and the DNA concentration was diluted to 1 ng/╬╝L using sterile water.
Polymerase chain reactions amplicon production and high-throughput sequencing
The V3ŌĆōV4 distinct regions of 16S rRNA (ribosome ribonucleic acid) genes were amplified using specific primers 515ŌĆ▓F (GTGBC AGCMGCCGCGGTAA) and 805R (GGACTACHVGGGTWT CTAAT) with barcodes [8]. The polymerase chain reactions (PCRs) were carried out in triplicate in a total volume of 25 ╬╝L containing 5 ╬╝M of each primer, 10 ng of DNA template, 4 ╬╝L 1├ŚFastPfu buffer, 2.5 mM dNTPs, and 0.4 ╬╝L of FastPfu polymerase (TransGen Biotech, Beijing, China). The following PCR cycles were used: initial denaturation at 94┬░C for 5 min, 30 cycles at 94┬░C for 50 s, 55┬░C for 30 s, and 72┬░C for 50 s, and a final extension at 72┬░C for 6 min. Then the mixture of PCR products was purified using the AxyPrep DNA Gel Extraction Kit (Axygen, Union City, CA, USA). Amplicons produced from different samples were sent to a commercial company (Majorbio, Shanghai, China) for sequencing on an Illumina HiSeq 2500 platform.
Analysis of short-chain fatty acids and pH
The four breeds pigsŌĆÖ fecal samples was mixed (1 g dry matter) with ten times Deionized Water. Then the mixture were centrifuged (10,000 rpm for 10 min) and 1,000 ╬╝L of the supernatant was added with 200 ╬╝L of crotonic acid (internal standard). Sample was injected on the gas chromatography-column and the content of SCFAs were analyzed using the method described by Yang et al [2].
Data analysis
First, the paired-end reads were assigned to samples based on their unique barcodes and truncated by removing the barcode and primer sequences. Paired-end reads were merged using FLASH (V1.2.7). The spliced sequences were called raw tags. Quality filtering of the raw tags was performed under specific filtering conditions to obtain high-quality clean tags according to the Qiime (V1.7.0) quality controlled process. The tags were compared with the reference database using the UCHIME algorithm to detect chimera sequences, and then the chimera sequences were removed to finally obtain the Effective Tags. Community analysis was performed using the Uparse software (v7.0.1001). Richness and diversity indices were produced using Mothur, and sets of sequences with Ōēź97% identity were defined as operational taxonomic units (OTUs). The representative sequences were distributed into phyla and genera according to the SILVA bacteria database (SILVA version 108, Silva, Bremen, Germany) following the Bayesian approach and using a cutoff of 97%. Subsequent analyses of alpha diversity and beta diversity were all performed basing on this output normalized data. Clusters were generated using a dissimilarity cutoff of 3% for calculating the Shannon, Chao 1 richness and abundance-based coverage estimators (ACE) using the Mothur package [9].
RESULTS
Bacterial diversity and composition of the four boar pig breeds
In this study, high-throughput sequencing technology was adopted, and a total of 194,813 valid sequence reads were generated from Duroc (47,796b), Hampshire (47,535b), Landrace (50,842a) and Yorkshire (48,640b). OTUs with a cut-off of 97% identity were then identified in Duroc (994ab), Hampshire (930b), Landrace (1,046a), and Yorkshire (1,028a) (Table 1). The Shannon diversity index, ACE and Chao1 were used to compare the bacterial diversities among the four boar pig breeds. Duroc (6.29a) exhibited the highest Shannon diversity index and had a more diverse bacterial community compared to Hampshire (6.20a), Landrace (6.08ab), and Yorkshire (5.85b). In addition, Hampshire contained a larger number of less-abundant bacteria than Landrace based on its greater Shannon index but lower ACE richness. The values of the Shannon and ACE indexes suggest that most of the fecal bacteria were captured (Table 1).
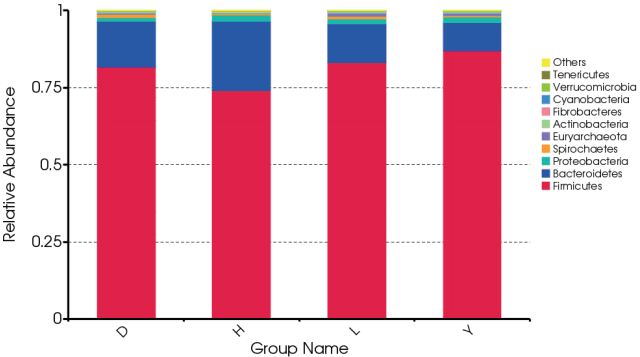
At the phylum level, Firmicutes and Bacteroidetes dominated the majority of the fecal microbiota regardless of breed. Other phyla were Proteobacteria, Spirochetes, Euryarchaeota, Actinobacteria, Fibrobacteres, Cyanobacteria, Verrucomicrobia, and Tenericutes. However, the bacterial community compositions of the breeds differed. For example, the proportion of the phylum Fimicutes was greater in the feces of Duroc (81.7%), Landrace (83.2%), and Yorkshire (87.0%) than in Hampshire (74.2%) (p = 0.064, 0.03, 0.003, respectively). By contrast, the proportion of the phylum Bacteroidetes was higher in the feces of Hampshire (22.2%) than in Duroc (14.8%), Landrace (12.5%), and Yorkshire (9.13%) (p = 0.065, 0.019, 0.002, respectively) (Figure 1).
At the class level, Clostridia, Bacilli, and Bacteroidia domi nated the majority of the fecal microbiota regardless of breed. Other abundant classes were Gammaproteobacteria, Spirochaetes, Methanobacteria, Erysipelotrichi, Coriobacteriia, Fibrobacteria, and Betaproteobacteria. However, differentially abundant bacterial communities were observed among the four breeds. For example, the proportion of Clostridia was much lower in the feces of Hampshire (52.3%) than in Duroc (59.9%), Landrace (66.1%), and Yorkshire (70.6%) (p = 0.021, 0.012, 0.001, respectively). The proportion of Bacteroidia was greater in the feces of Hampshire (22.2%) than in Duroc (14.8%), Landrace (12.5%), and Yorkshire (9.13%) (p = 0.065, 0.019, 0.002, respectively) (Supplementary Figure S1).
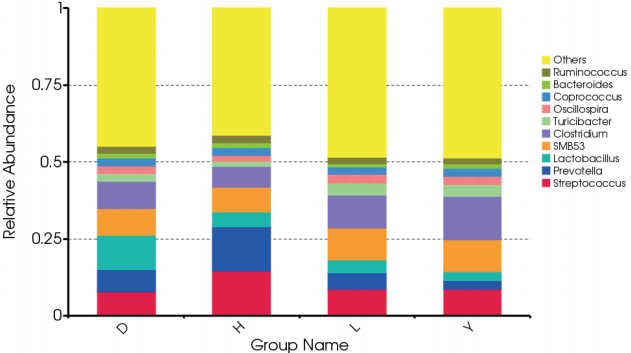
At the genus level, we calculated the most abundant nine genera in the fecal microbiota of all breeds of pigs as core bacteria that accounted for 44.82% of the total sequences (Figure 2; Supplementary Figure S2). Most of the core genera were Firmicutes, except Prevotella and Bacteroides, which belong to Bacteroidetes. Similar to a previous report, Prevotella and Streptococcus were the most abundant genera [10]. Streptococcus (14.6%) and Prevotella (14.3%) were more abundant in the fecal microbiota of Hampshire pigs than the other breeds (p<0.05). A previous study showed that Prevotella decreased from 30% of all bacteria to 4.0% as pigs aged; these bacteria degrade mucin and xylan, which might be helpful in feed digestion [11,12]. The abundance of the genus Lactobacillus (11.1%) was markedly greater in Duroc pigs than in the other three breeds (p<0.05), in contrast to a previous study that observed that the abundance of Lactobacillus was highest in Landrace pigs [7]. The abundance of the Clostridium genus differed among the four breeds and was highest in Yorkshire (14.2%) and lowest in Hampshire (6.67%, p<0.05). The Turicibacter genus was more abundant in both Landrace (3.81%) and Yorkshire (3.87%, p<0.05) than the other two breeds. The Oscillospira genus was more abundant in Landrace (3.07%) but less abundant in Hampshire (2.01%, p<0.05). The Coprococcus genus was more abundant in Yorkshire (2.57%) but less abundant in Landrace (2.21%, p< 0.05). Bacteroides was less abundant in Landrace whereas Ruminococcus was more abundant in Hampshire, but these differences were not significant.
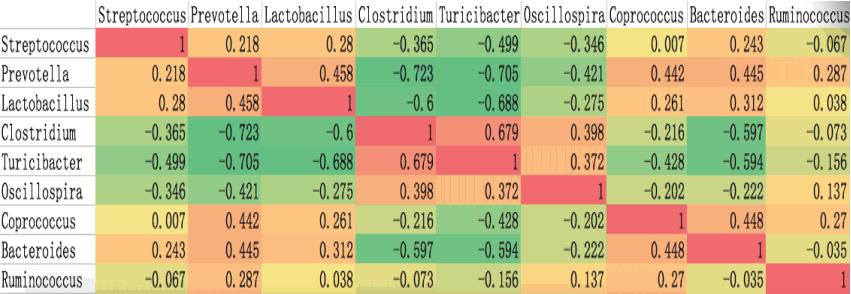
SpearmanŌĆÖs rank correlation test was used to analyze the co- occurrence patterns among the nine core bacteria (Figure 3). Some bacteria were positively correlated with each other. For example, Prevotella and Lactobacillus were significantly correlated with each other (rho = 0.458) and with other bacteria such as Coprococcus (rho = 0.442, 0.261), Bacteroides (rho = 0.445, 0.312) and Streptococcus (rho = 0.218, 0.28). Clostridium was positively correlated with Turicibacter (rho = 0.679) but negatively correlated with the other six bacteria. In contrast to previous findings for human gut bacteria [13], co-occurrence between Prevotella and Ruminococcus was observed in the four pig breeds (rho = 0.287), and we observed co-exclusion of Prevotella and Clostridium (rho = ŌłÆ0.723), Turicibacter (rho = ŌłÆ0.705) and Oscillospira (rho = ŌłÆ0.421). As shown in Figure 5, Streptococcus, Prevotella and Lactobacillus co-occurred and cooperated to suppress three other cooperative genera: Clostridium, Turicibacter, and Oscillospira. These results may reflect multiple mechanisms such as secretory products and competition for nutrients [14]. The contribution of these co-occurring genera and co-excluding genera as revealed in this study may have some fundamentally important function and must be researched deeply.
SCFAs, pH, and cellulose activity of the fecal microbiota of the four pig breeds
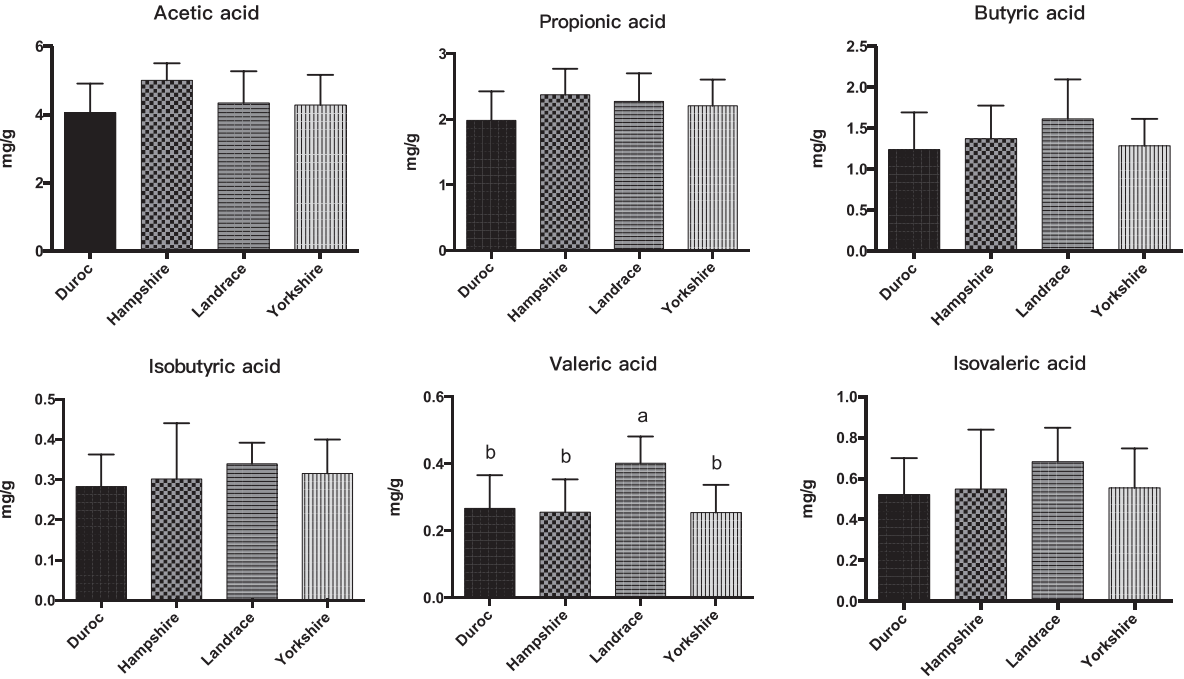
We also measured the SCFAs, pH, and cellulose activity of the fecal microbiota of the four breeds of pig (Figures 4, 5). The butyric, isobutyric, valeric, and isovaleric acid levels were higher in Landrace pigs than in the other three breeds, whereas acetic and propionic acid were highest in the Hampshire breed. These differences were associated with pH: pH was significantly lower for Hampshire and Landrace than for the other two breeds. Most mammals have a limited ability to produce cellulase and thus cannot digest fibers by themselves. Some gut bacteria of pigs, such as Clostridium, Bacillus, Ruminococcus, and Bacteroides, produce cellulases to help their host digest diet fibers.
Different abundant bacteria and principal coordinate analysis of the four pig breeds
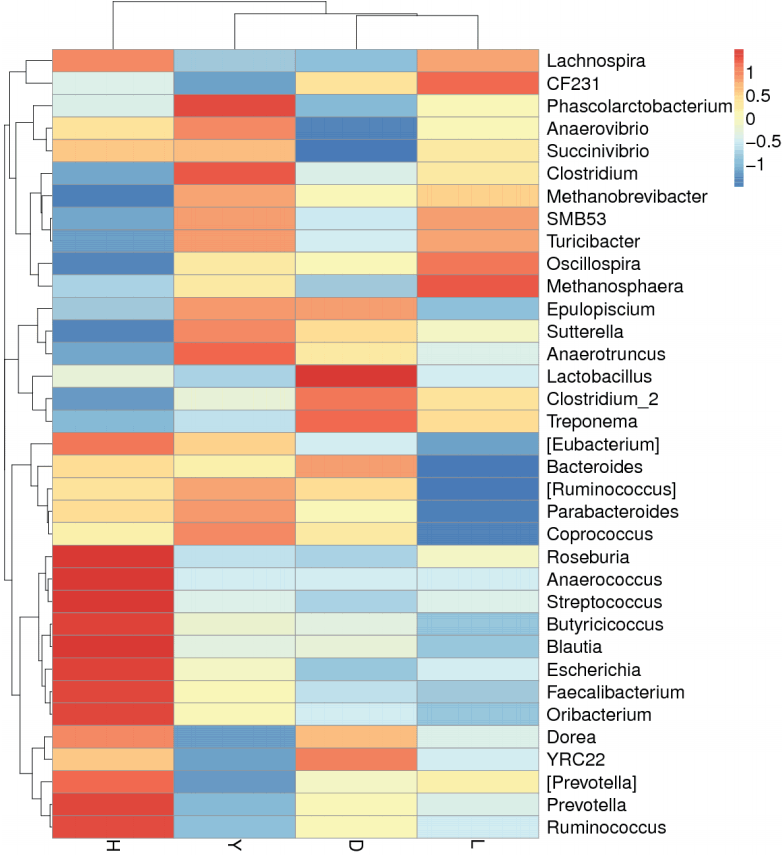
The abundance distribution of the dominant 35 genera among the four breeds was displayed in a species abundance heatmap (Figure 6). The information for the clustering results of the samples and taxa revealed whether the samples with similar processing were clustered or not and the similarity and difference between the samples. Although these 35 genera were common to the fecal microbiota of all of the pig breeds, the heatmap revealed several genera that exhibited variations between breeds. For example, the genera Streptococcus, Prevotella, Ruminococcus, Roseburia, Anaerococcus, Butyricicoccus, Blautia, Escherichia, Faecalibacterium, and Oribacterium were more abundant in Hampshire pig fecal microbiota. The genera Phascolarctobacterium and Clostridium were more abundant in Yorkshire. The genera Lactobacillus and Treponema were more abundant in Duroc, whereas Methanosphaera was more abundant in Landrace. Thus, the variations among the breeds were specific and functionally relevant.
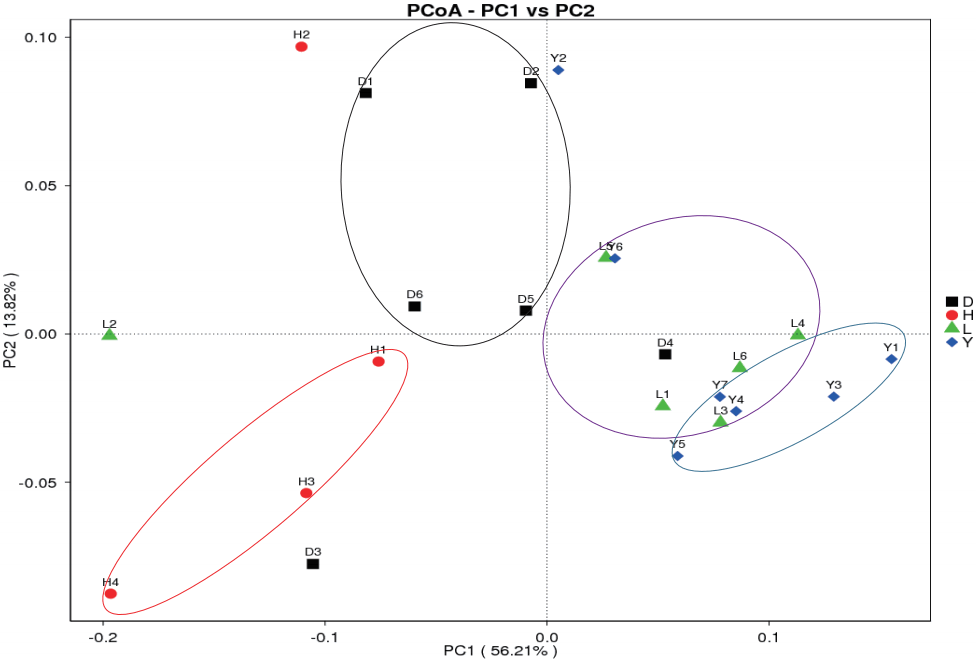
Principal coordinate analysis (PCoA) is an ordination tech nique that is similar to PCA. PCoA identifies the main elements and structure from reduced multi-dimensional data series of eigenvalues and eigenvectors. The advantage of PCoA over PCA is that each ecological distance can be investigated. PCA identifies the main coordinates based on the similarity coefficient matrix of all samples, whereas PCoA is based on a distance matrix. Weighted UniFrac and Unweighted UniFrac are calculated to assist the PCoA analysis. Clustered samples represent high species composition similarity compared to separated samples. The fecal bacterial communities clustered according to different breeds (Figure 7). The microbiomes of the Landrace and Yorkshire breeds had high similarity but were clearly separated from those of the Duroc and Hampshire breeds, similar to a previous result [7].
According to the analysis results for OTU clustering and the research requirements, we normalized the OTU table and analyzed both the shared and unique information for different samples (groups). The Venn diagram (Supplementary Figure S4) revealed that the four breeds shared most OTUs (783), but some breed-specific OTUs were observed. For example, Duroc had 16 specific OTUs, Landrace had 41, Yorkshire had 27, and Hampshire had 11. The associations of these specific OTUs with pig genetics and gut ecology warrant further research.
DISCUSSION
Previous studies have suggested that pig breed affects the composition of Firmicutes, Bacteroidetes, and sulfate-reducing bacteria, which are higher in Chinese native pig breeds than in foreign breeds [2]. Both genetic and cage effects are significantly associated with microbiota composition [15]. Firmicutes and Bacteroidetes were abundant phyla in the fecal bacterial community at pre-weaning [16], post-weaning and four years of age [12] but changed as pigs aged. Firmicutes and Actinobacteria are more abundant in lean Gottingen minipigs than in lean Ossabaw minipigs [17]. Firmicutes and Bacteroidetes are also the two most abundant phyla in the healthy human gut microbiota, but the ratio of these two phyla varies among individuals [4,18]. A previous study reported similar results for the gut tract of other breeds of pig [12].
At class level, the result differs from the results of previous studies in which the dominant class was Bacteroidia followed by Clostridia for all breeds [1,7]. In addition, the proportion of Bacilli was greater in the feces of Duroc (21.3%) and Hampshire (21.2%) than in Landrace (16.7%) and Yorkshire (15.8%). A previous study had showed that Bacilli and Clostridia were more abundant in obese Ossabaw minipigs, whereas Bacteroidia was more abundant in lean Ossabaw minipigs [17].
The differences in the abundance of genera among various breeds may be influenced by differences in gut function among specific breeds [19]. Functional analysis of Yorkshire pig fecal DNA extracts revealed that carbohydrate metabolism-associated bacteria reach 13% of the swine metagenome [11]. A previous study found that the genera Prevotella and Bacteroides were more abundant in obese Gottingen minipigs, whereas Clostridium was higher in lean Gottingen minipigs. However, another study reported that Prevotella had a greater abundance in lean Ossabaw minipigs, whereas Clostridium was more abundant in obese Ossabaw minipigs [17]. Similar clustering of bacteria in pigs of the same age was observed in different breeds of pigs [12]. Oscillospira, an enigmatic bacterial genus that has never been cultured and is positively correlated with leanness and health, has been detected frequently in the human gut [20]. From the SpearmanŌĆÖs rank correlation, a perfect positive correlation indicates that these bacteria cooperated with each other, whereas perfect negative correlation represents suppression. SpearmanŌĆÖs rank correlation provides valuable and complementary information.
Host genetics have an important influence on microbial di versity [19] and breed-specific bacteria [7]. Bacillus subtilis BY-2, which produces cellulose, has been isolated from the intestine of Tibetan pigs whose diet consists of forage grass [21], suggesting that the function of gut bacteria depends on the genome of the host breed. However, a deeper level of sequencing data is required to determine the specific metabolic functions that are conserved.
Previous studies have demonstrated that Landrace and York shire are more genetically similar, with closer fecal microbiomes, whereas Duroc and Hampshire are genetically distant [7,22]. The microbial compositions of the Duroc, Landrace, and Yorkshire breeds were clustered in one group and differed from those of Chinese breeds such as Bama and Meishan sows [2]. The microbial communities varied between Gottingen and Ossabaw minipigs, which are models of obesity [17]. Research on mice has demonstrated that genetic distance correlates positively with microbiota distance; genetically similar mice have more similar bacterial compositions than do genetically distant mice [15]. A study of different Chinese ethnic groups indicated that Mongol, Tibetan, Zhuang, Han, Bai, Kazakh, and Uyghur ethnic groups were separated from each other significantly (p = 0.0001) [4].
SCFAs, including acetate, propionate, butyrate and pentanoic acid, are major anions in the gut and are absorbed rapidly by colonic epithelial cells. Acetate can enter systemic circulation and be used for lipogenesis; butyrate is the major energy source for colonocytes [23]. These small anions can improve gut barrier function and are associated with many metabolic activities [24]. These small molecules can also exhibit anti-inflammatory effects and protect the host against gut diseases [25,26]. Surprisingly, eight core bacteria exhibited potential for SCFA production. Bacteria such as Bacteroides can produce acetate and succinate [27]. Clostridium, Ruminococcus, and Coprococcus are candidate genera associated with butyrate production [10,28]. Streptococcus, which belongs to the order Lactobacillales, can produce acids. Prevotella has an essential role in metabolizing plant cell wall dietary fiber and thus can produce significant amounts of SCFAs [29], consistent with our findings. Prevotella was positively correlated with five core bacteria that all produce SCFAs and are believed to help pigs better adapt to a diet with fiber, which may explain the higher cellulase activity of Hampshire pigs compared with other breeds with a lower abundance of Prevotella. As discussed previously [20], Oscillospira probably produces SCFAs and is negatively associated with inflammatory diseases and body mass index; thus, this genus has been linked to host leanness. The genus Lactobacillus can produce lactic and other acids such as SCFAs and therefore exhibits great importance for host health [2]. However, these bacterial genera require extensive research for both genomic and functional characterization.
CONCLUSION
Overall, we first compared the fecal microbiota of the four breeds of boar pig by high-throughput sequencing and identified nine core bacteria that may contribute to host digestion and health. Among the nine bacteria, eight genera had the ability to produce SCFAs, and some bacteria can secrete cellulase to aid host fiber digestion. PCoA revealed that Hampshire was separated from the other three breeds, whereas the bacteria of Landrace and Yorkshire were more closely related to each other. SpearmanŌĆÖs rank provided valuable information about bacterial competition and cooperation. Several breed-specific bacteria were identified that are likely associated with host genetics and characteristics, but further studies are needed from the physiological perspectives of the host.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







