





INTRODUCTION
China produces more than 50 million tons of pork each year, accounting for over 50% of the total global production [1]. Even a slight increase in Chinese pork production will have a significant impact on the global pork market. Reproductive traits, such as total number born (TNB) and number born alive (NBA), have been considered as the most important index included in the selection indices of pig breeding programs for evaluating sow productivity [2]. Up to the present, selection based on traditional breeding methods using best linear unbiased prediction has been successful in improving maternal reproductive traits [3]. However, the genetic architecture of reproductive traits is very complicated due to low heritability, minor genes, maternal effects and environmental factors [4], resulting in the difficulty deciphering the genetic architecture of reproduction traits. Over the past 20 years, the dense genome coverage provided by high-throughput chip genotyping makes it possible to exploit the linkage disequilibrium (LD) between single nucleotide polymorphisms (SNPs) and quantitative trait locus (QTL) through genome-wide association study (GWAS) to identify genes related to traits of interest. Several GWAS have been revealed significant associations for economically important traits such as reproduction traits [5], growth traits [6], meat traits [7], and feed conversion [8].
Exploring the loci and genes affecting sow reproduction performance is necessary for understanding the genetic characteristics of these traits and increasing the speed of genetic improvement [9]. On one hand, genes such as estrogen receptor 1 [10], insulin-like growth factor 2 [11], and aryl hydrocarbon receptor [12] had been identified as important candidate genes positively associated with reproduction. But these genes explain only a relatively small proportion of the genetic variance. On the other hand, limited by the size of the population and other factors, only small number of genes were detected in association analysis. According to the pig QTLdb (https://www.animalgenome.org/cgi-bin/QTLdb/SS/index), until now, there are 2,135 QTLs reported for overall traits linked to reproduction including 6 QTLs for endocrine, 1,013 for litter traits, 668 for reproductive organs, and 448 QTLs for reproductive traits. Among them, 522 QTLs identified for TNB (350 QTLs) and NBA (172 QTLs) compared with 8,962 QTLs for fatness or 1,745 for growth traits.
Most of those studies mainly utilize one pure breed or an intercross population, with the result only reflecting one specific breed’s linkage disequilibrium character. As the most popular commercial pig breed, Yorkshire is usually used as terminal dam line in pig hybrid production, and the improvement on their reproduction traits is therefore very important. There were two main objectives in our study. The first one was to detect significant SNPs and candidate genes in four populations from different genetic backgrounds using conventional single-trait GWAS respectively. The second one is to improve the power of GWAS by enlarging population size through implementing a meta-analysis for multiple traits within a population or for same trait across populations.
MATERIALS AND METHODS
Ethics statement
The whole procedure for collecting ear tissue samples was carried out in strict accordance with the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the China Agricultural University. The IACUC of the China Agricultural University specifically approved this study (permit number DK996).
Animals and phenotype
A total of 3,121 Yorkshire pigs used in this study were sampled from four pig breeding farms (abbreviated as LM, FJ, XD, ZX for convenience), including 2,247 progeny of three different American Yorkshire populations and 874 progeny of one British Yorkshire population (XD). Animals from LM and ZX are descendants of American Yorkshires but from different breeding companies, while LM and FJ came from the same breeding companies. The progeny of American Yorkshires were born in 2013 through 2018 and came from 222 sire families (8 to 76 offspring in each family with an average of 15), and the progeny of British Yorkshires were born in 2007 through 2013 and came from 129 sire families (10 to 71 offspring in each family with an average of 7). There was no genetic connectedness between LM, XD, and ZX according to the pedigree information. Phenotypic records included two reproductive traits, TNB and NBA. The populations and phenotypes information are presented in Table 1.
Breeding values for NBA and TNB were routinely estimated by the breeding companies using a standard animal repeatability model which was separately implemented in each population, and were obtained from the National Swine Genetic Improvement Center of China (http://cnsge.nahs.org.cn/); afterwards, corrected phenotypic values were calculated as EBV plus the estimated residual for each individual in each population.
Genotyping and quality control
Genomic DNA was extracted from blood samples using a TIANamp Blood DNA Kit (catalog number DP348; Tiangen, Beijing, China). Genotyping was performed using a PorcineSNP80 BeadChip (Illumina, San Diego, CA, USA), which includes 68,528 SNP across the entire pig genome. Genotype quality control was carried out using PLINK 1.9 software [13] separately for each population. First, individuals with call rates (CR) less than 90% were removed and then SNP with CR less than 90%, minor allele frequencies <3%, or significant deviation from the Hardy–Weinberg equilibrium (p<10× 10−6) were removed. After genotype quality control, 3,121 individuals and 49,839 SNP remained for further analysis.
Population structure
Because the genetic backgrounds of four Yorkshire populations in this study are different, a principal component analysis (PCA) was carried out to detect the population stratification using GCTA software [14]. In order to keep the independence of SNPs, the adjacent SNPs with r2 greater than 0.2 were further pruned after genotype quality control, and in total 29,229 SNPs were used in PCA. The linkage disequilibrium within each population was calculated using PLINK software [13] as well. Meanwhile, a quantile-quantile (Q-Q) plot was generated to assess the influence of population stratification on the GWAS.
Statistical analysis
Single-population GWAS through linear mixed model was carried out in each pig population separately. Based on single-population analysis, the meta-analysis within population and cross populations were conducted, respectively.
Genome-wide association study for a single trait in a single-population
Linear mixed model
A linear mixed model was implemented to detect the association of SNP with growth and fatness traits. The model in this study is a single SNP regression model. The model includes a random polygenic effect to account for shared genetic effects of related individuals and to control population stratification. The statistical model is described below:
in which yc is the vector of phenotypes (corrected phenotypic values); 1 is a vector of ones; μ is the overall mean; b is the average effect of the gene substitution of a particular SNP; x is a vector of the SNP genotype (coded as 0, 1, or 2); g is a vector of random polygenic effects with a normal distribution g ~ N(0, Gσa2), in which σa2 is the polygenic variance and G is the genomic additive relationship matrix and was constructed using all markers following VanRaden [15]; Z is an incidence matrix relating phenotypes to the corresponding random polygenic effects; and e is a vector of residual effects with a normal distribution N(0, Iσe2), in which σe2 is the residual variance. The software GCTA [14] was used to fit the model.
Afterwards, Bonferroni correction at a significance level of 0.05 was used to identify significant SNP. There were 52,173, 52,804, 52,526, and 52,267 qualified SNPs in the four populations (LM, FJ, XD, and ZX), respectively. The p values of the 5% genome-wide and suggestive significant thresholds were equal to 0.05/SNPs number and 1/SNPs number, respectively, in four populations.
Meta-analysis of GWAS for a single trait across populations (MS-GWAS)
Based on the results of GWAS separately in four populations through single-population analysis, and the meta-analysis based on Fisher’s method was carried out to combine P-value probabilities from each test into one test statistic (X2) using the formula:
in which pi is the raw p-value of tth study for t = 1, …, T, in which T is the number of independent studies. When all the null hypotheses are true, this combined test statistic follows a χ2 distribution with 2T of degree of freedom. Therefore, the new p-value from the meta-analysis was calculated using:
in which χ22T is a χ2 variable with 2T of degree of freedom. In our study, we used the common SNP in four population by Fisher’s method to calculate a meta-analysis p-value. Afterwards, Bonferroni correction at a significance level of 0.05 was used to identify significant SNP. There were 48,966 common SNPs in the four populations, the threshold p-value for each SNP at significance level of 0.05 was 1.02×10−6 (0.05/48,966).
Meta-analysis of GWAS for multiple traits within population
In the present study, the traits NBA and TNB reflecting similar fertility function can be considered as different traits with some common genetic components. Therefore, the meta-analysis was performed within reproduction traits, TNB and NBA.
An approximated chi-square statistic [16] was applied to test whether there is at least one of the SNP effect of studied traits was not equal to zero. For each SNP, chi-square statistic of a multi-trait meta-analysis was calculated using the following formula:
where ti was a vector of signed t-values of the ith SNP for all studied traits,
t i ′
Identification of candidate genes
To identify functionally plausible candidate genes near the significant SNP, the genes located in or overlapping the region between the 0.5 Mb upstream and 0.5 Mb downstream of the significant SNP were obtained using Ensemble (http://www.ensembl.org/Sus_scrofa/Info/Index; Sscrofa 11.1 genome version). Gene ontology analysis was carried out using the DAVID bioinformatics resource (https://david.ncifcrf.gov/). Pathway analysis was conducted using the online KEGG (http://www.kegg.jp/kegg/pathway.html) and GeneCards (http://www.genecards.org/) tools.
RESULTS
Population structure
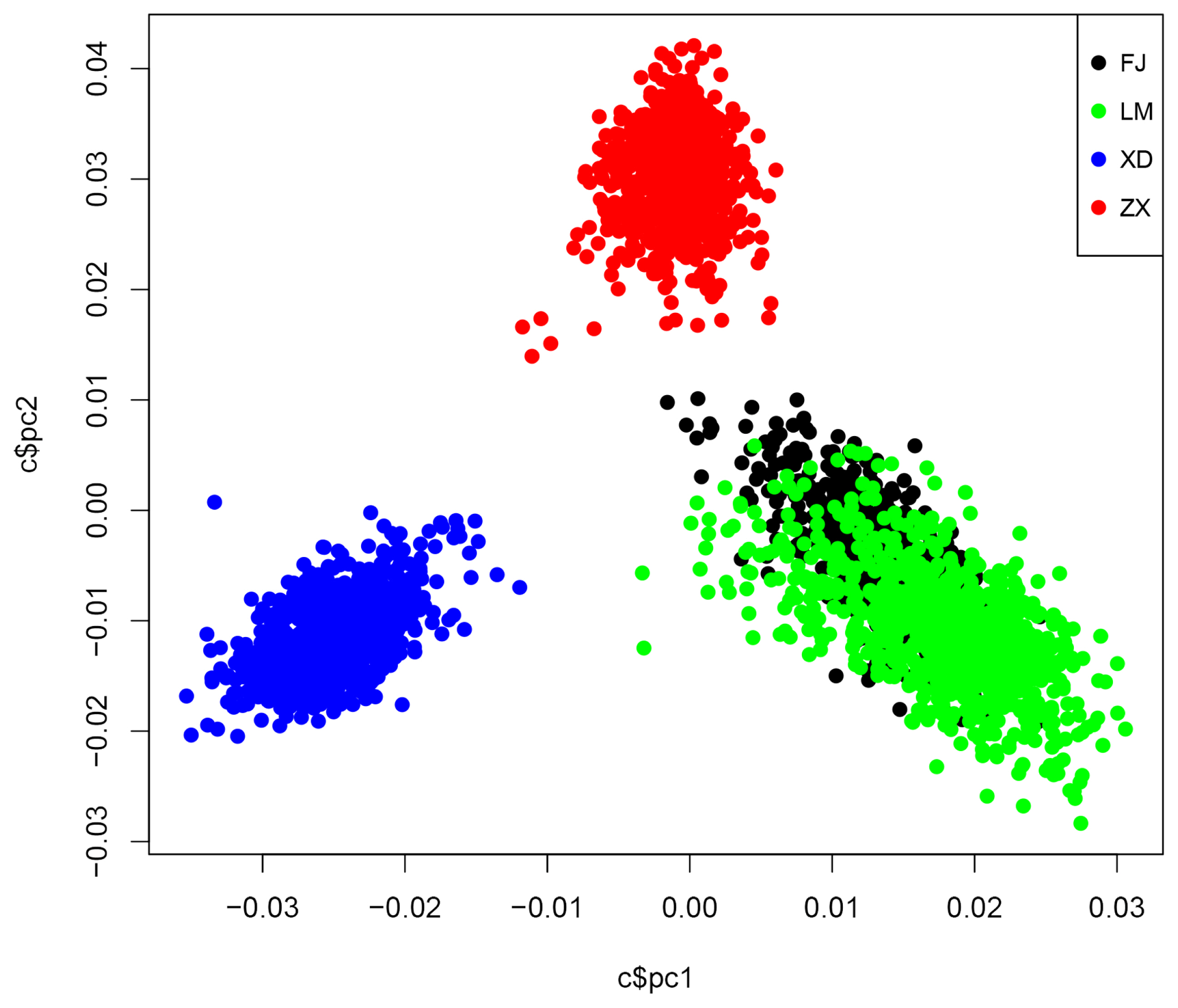
To identify the population structure of the four Yorkshire populations involved in this study, a PCA was performed using the chip data. As shown in Figure 1, the four Yorkshire populations from four farms can be clearly identified through PCA. The genetic backgrounds of the LM and FJ populations were classified nearly into one cluster implying no significant genetic differentiation among them. Meanwhile, both LM and FJ populations were divergent from ZX population, as they came from different American Yorkshires breeding companies. Likewise, XD was distantly related to FJ, LM, and ZX due to its British origins.
SNPs identified by SS-GWAS for TNB and NBA in four populations
All significant SNPs associated with TNB and NBA traits in single population analysis are illustrated in Table 1. For SS-GWAS, the p values of the 5% (suggestive) genome-wide significant threshold were equal to 9.58×10−7 (1.92×10−5), 9.47×10−7 (1.89×10−5), 9.52×10−7 (1.90×10−5), 9.57×10−7 (1.91 ×10−5) in these four populations (LM, FJ, XD, and ZX), respectively. A total of 13 SNPs, of which six SNPs reached the genome-wide suggestive level for NBA and seven SNPs reached the genome-wide suggestive level for TNB, as shown in Table 2. Among that, six suggestive SNPs for XD, one for FJ, six for ZX and no SNPs for LM. The results of single population GWAS are in Supplementary Figure S1. No significant SNP was found in the single population GWAS results.
SNPs identified by MS-GWAS for NBA and TNB across populations
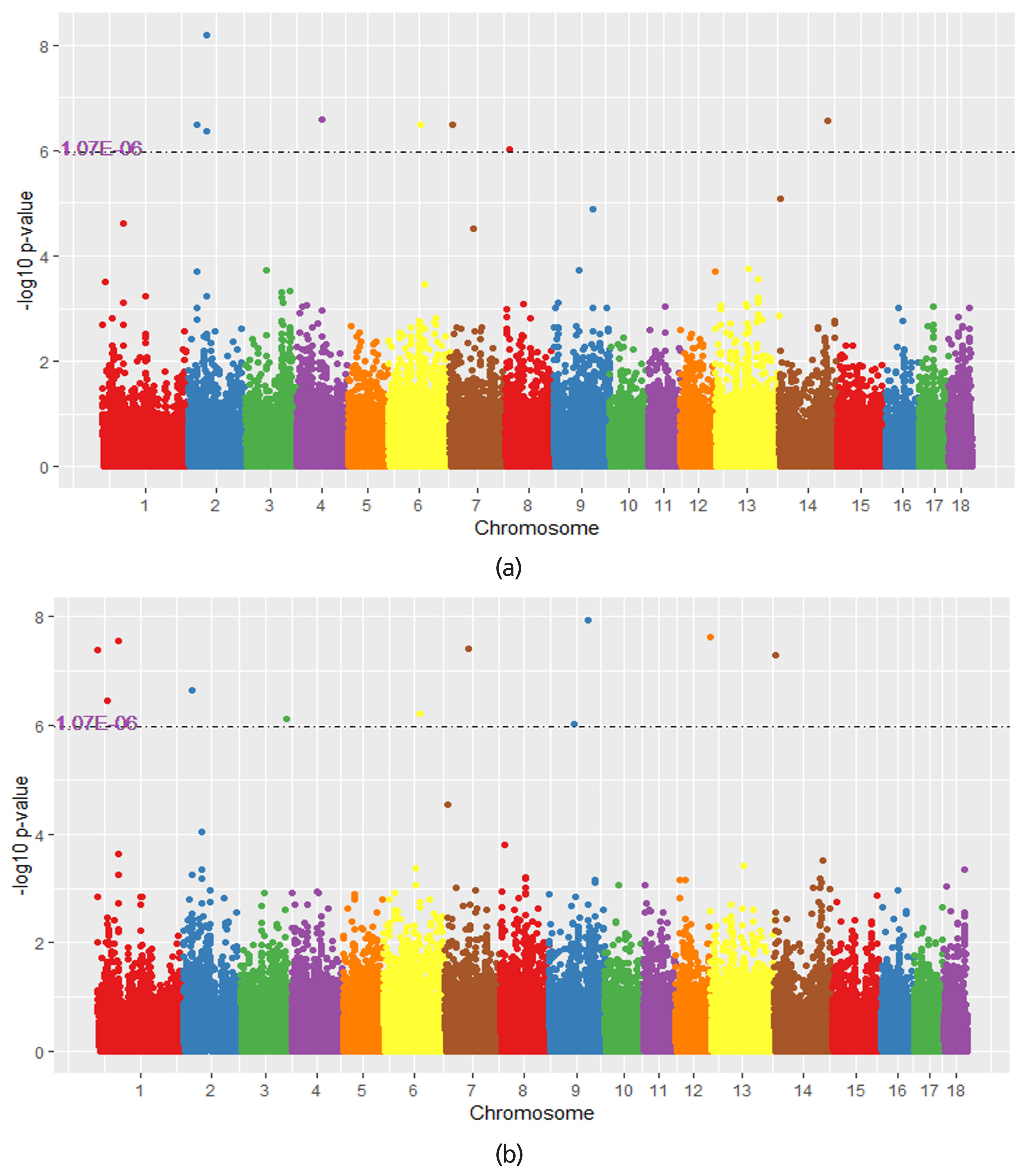
Manhattan plots for meta-analysis across populations are presented in Figure 2, while the summary of significant SNPs for TNB and NBA in the meta-analysis across populations is listed in Table 3. In total, 19 significant SNPs were detected for the traits analyzed in the meta-analysis across populations: 11 for TNB and 8 for NBA. Among them, 5 significant SNPs which were detected in TNA or NBA were also reached a suggestive level in another trait. In addition, the number of significant SNPs identified in meta analysis across populations were larger and more significant than in single-trait analysis. Ten suggestive SNPs which detected in SS-GWAS reached the higher level of significance in meta analysis across populations. Besides that, 11 SNPs that were significant in meta analysis across populations became more significant in multi-traits meta in the next analysis.
SNPs identified by MM-GWAS within population
Genomic correlation matrices between TNB and NBA were constructed in four populations, respectively. The absolute of genomic correlation was 0.89, 0.85, 0.87, and 0.89 for each population of LM, FJ, XD, and ZX, respectively. Manhattan plot for MM-GWAS within four populations are presented in Figure 3. In LM population (Figure 3a), the MM-GWAS revealed 42 SNPs associated with both TNB and NBA, including 13 with genome-wide significance and 29 with suggestive significant level. There were 60 SNPs identified for FJ population (Figure 3b), including 13 genome-wide significant loci and 47 genome-wide suggestive loci. For XD population (Figure 3c), 24 reached the significant level and 42 reached suggestive significant level. For ZX population (Figure 3d), 16 significant and 35 suggestive loci were identified. Compared the results of four populations, only 11 SNPs reached the significant or suggestive significant level in each population (Table 4). Besides that, these 11 significant/suggestive SNPs were all overlapped in the result of meta-analysis of GWAS (MS-GWAS). The significant/suggestive SNPs detected using the multi-trait meta-analysis within population are listed in Table 4.
Identification of candidate genes
Based on 11 common significant SNPs associated with TNB and NBA identified by two meta-analysis methods, 11 genes which located within the region between the 0.5 Mb upstream and 0.5 Mb downstream of the significant/suggestive SNP were found and annotated (Tables 2 to 4). While Go ontology analysis revealed that, there were five annotated genes had a highlight biology function with TNB and NBA. All these annotated genes were selected based on the Sus scrofa 11.1 genome assembly. Further function annotation was carried on based on the NCBI database (https://www.ncbi.nlm.nih.gov/).
Quantitative traits locus overlapped with SNPs
Until now, there were 522 pig QTLs identified for reproduction traits TNB (350 QTLs) and NBA (172 QTLs) in the pig QTL database (https://www.animalgenome.org/cgi-bin/QTLdb/SS/index). After comparing these QTL with the regions of 11 common significant SNPs, 6 SNPs were identified located in 5 QTLs which were identified before in Sus scrofa chromosome 6 (SSC6), SSC7, and SSC9 for TNB and NBA traits (https://www.animalgenome.org/cgi-bin/QTLdb/SS/traitmap?trait_ID=157 or trait_ID=156). This implies the functional genes such as msh homeobox 1 (MSX1), vascular endothelial growth factor A (VEGFA), forkhead box Q1 (FOXQ1), and LHFPL tetraspan submily member 3 (LHFPL3) around these SNPs are likely candidates for TNB and NBA traits.
DISCUSSION
According to the results of genes annotated, a total of 11 genes of which five are relevant with two traits of reproduction, these candidate genes could regulate or influence TNB and NBA through different kinds of biological processes and pathways. MSX1, spindlin 1 (SPIN1), VEGFA, FOXQ1, and LHFPL3 are highlighted as promising biological candidate genes for reproduction traits. Except MSX1 and VEGFA, SPIN1, FOXQ1, and LHFPL3 are reported as related to reproduction in pigs for the first time. MSX1 is associated with the utero embryonic development and some classical pathways in embryo development such as Wnt/Hedgehog/Notch pathway. Daikoku et al [17] and Nallasamy et al [18] found that Msx1 was expressed in the preimplantation mouse uterus, and are critical for fertility in mice. Cha et al’s research [19] suggested that these transcription factors have cell-specific functions in the pregnant uterus, and its subsequent morphological and functional changes. MSX1 had been already reported relevant with NBA and TNB in pigs [20]. SPIN1 is necessary for normal meiotic progression in mammals [21]. In a previous study, Choi et al [22] revealed that SPIN1 may play an important role in meiosis II (MII) arrest as well as in the regulation of early embryonic development. As to VEGFA, has key function in uteroplacental vasculogenesis during embryonic implantation and provides the vascular network to the placenta [23]. In previous pig GWAS studies, the VEGFA gene has been reported to be associated with TNB and NBA [20,24], and our study confirmed the previous investigations. Also, LHFPL3, a member of the family of LHFP-like genes. A direct link between LHFPL3 and reproductive traits has not been reported before, but Ptacek et al [25] reported that it was closely related to uterine leiomyoma and highly expressed in the uterus. As we all known, uterus is important to embryonic development. So, LHFPL3 might have a key function in conceiving and maintaining pregnancy. FOXQ1 has not been defined in pigs, whereas in mice, it is involved in patterning the early embryonic mesoderm [26] and expressed at embryo day 8.5 [27]. Our findings will be helpful for a better understanding of the role of FOXQ1 for embryo development in pig reproduction.
Population stratification is a major factor in false positives in GWAS for significant SNPs [28]. At present, there are 4 popular methods to resolve the problem of population stratification, genomic control, structured association, PCA, and linear mixed model which can deal with population stratification by taking polygenic effects into account. In general, a genomic inflation factor λ of <1.05 indicates no population stratification [29]. Our values were 1.788 to 3.24 for TNB and NBA in four populations before using genomic relationship matrix G. According to Janss et al [30], inclusion of principle components in model to account for population structure is not required, mainly because of the genetic variation accounted for after the genomic relationship matrix G has been considered in the model. Thus, PCA is incorporated in individual population analyses. Here, population stratification was adjusted through a constructed relationship matrix using genotype data in the linear mixed model. Genomic relationship can more accurately reflect the actual existing relationship between animals compared to pedigree-based relationships. By using G matrix (genomic additive relationship matrix) instead of A matrix (additive genetic relationship matrix), genomic inflation factor λ reduced to 1.02 to 1.04 for TNB and NBA which implied the genomic relationship matrix adequately controlled population stratification, as the Q-Q plot indicated (Supplementary Figure S2). This made the GWAS results more reliable.
In our study, two meta-analysis were conducted to explore the genomic loci for TNB and NBA based on the results of single trait GWAS in four pig populations. The results of GWAS for a single trait in a single population showed that no common significant SNPs were detected in the four populations (Table 1, Supplementary Figure S1) for TNB and NBA. Most of suggestive SNPs detected in XD did not appear in the ZX population. Only two SNPs (WU_10.2_4_80076056 and WU_10.2_1_11153176) were repeated in FJ and XD. These observations further reflected the complex genetic architecture of pig reproduction traits. The low consistency of findings from single population is similar with other investigations in GWAS. Many researches [31] performed a GWAS on different pig populations and revealed SNPs and candidate genes related target traits. But no overlapped significant results were identified in multiple populations. A possible reason for lack of genome-wide significant SNP is that small sample size, different population structures or the complexity of traits [32]. Liu’s et al research [33] confirmed that a large population size is important for GWAS in traits with low heritability. Combining different populations could reveal hidden or unclear associations that may not be detected by an independent study [32]. Novel significant SNPs could be detected in GWAS due to the greater power with increased sample size [34]. Our results indicated that meta-analysis could be efficient tool to improve the power of GWAS by combining different populations. Moreover, meta-analysis can increase statistical power especially for a locus with small effect by collectively using information from multiple independent studies [34].
No locus was identified significant by single population GWAS for TNB and NBA, with most significant SNPs detected across the two meta analysis approaches. In addition, compared to GWAS, 11 novel significant SNPs were detected using meta-analysis. Besides that, meta-analysis made the p-value smaller and more significant which were also consistent with Guo et al [35] and Le et al [36].
Significant SNPs were not found in single population GWAS, which showed the enhanced capacity of multi-trait approaches for detecting SNPs, particularly when the phenotypes are correlated [37]. Our finding that SNPs, which cannot be detected at a genome-wide significance level in GWAS, can be uncovered in a multi-trait approach corroborates the findings of Willer et al [32]. The joint association analysis of multiple phenotypes might be a more powerful approach to detect SNPs that underlie correlated traits than the multi-trait test statistic applied in our study [38].
In summary, we conducted GWAS separately in four Yorkshire populations, which have distinct genetic backgrounds, to identify genomic loci for reproduction traits of TNB and NBA. Based on results of GWAS, two kinds of Meta-analysis were implemented to improve the power of gene identification. Compared to the single population GWAS, single-trait meta analysis can improve the detection power to identify SNPs in a larger multiple population. The multiple-trait analysis reduced the power to detect trait-specific loci but enhanced the power to identify the common loci across traits. In total, 11 significant loci were identified associated with target traits. SPIN1, VEGFA, FOXQ1, MSX1, and LHFPL3 are five functionally plausible candidate genes for TNB and NBA. Our findings further revealed that the meta-analysis and the multi-trait method can be used to increase the power of GWAS to identify genes relevant with important traits of interest.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement
Supplement Print
Print







