




INTRODUCTION
Many cattle breeds have been domesticated for milk or beef all over the world since Neolithic age. Ancient populations used cattle breeds of Bos taurus (non-humped) or Bos indicus (humped), mainly for draught work, clothes, meat and manures. After the industrial revolution, the cattle breeds were intensively selected for meat protein. Based on the geographical distribution, native or local breeds can be classified as for either meat or dairy purposes, based on such characteristics as body size, milking potential, age at puberty, temperature adaptability, fleshing ability, muscle expression, cutability, and marbling.
Hanwoo (Bos taurus coreana) is one of the economically and biologically important ruminants in South Korea. Hence, characterization of cattle genomes may afford an opportunity to gain a deeper understanding of particular breed genetics (Decker et al., 2009). Korean beef consumers prefer Hanwoo meat rather than imported meat, because of is high intramuscular fat (IMF or marbling), flavor and tenderness. However, large volumes of imported beef are gradually occupying Korean beef market, due to the competitive beef price and FTA (free trade agreement) (Paredi et al., 2012). The Hanwoo breed has characteristic yellow hair and doesnŌĆÖt have more than two colors. Besides, Hanwoo is known to be strongly disease-resistant, implying that a distinct level of activation exists in their immune-related pathway (Kim et al., 2013).
Single nucleotide polymorphism (SNP) and copy number variation are the most abundant and common type of genetic variations existing in mammalian genomes. High-throughput genotyping techniques have allowed the use of molecular marker sets to identify the evolution, imprints of domestication and breed development of many species, including humans (Novembre and Ramachandran, 2011), sheep (Kijas et al., 2009), pigs (Groenen et al., 2012), dogs (Vonholdt et al., 2010) and horses (McCue et al., 2012). In Hanwoo, association studies between genes and meat quality have been reported and also genetic information of the cattle genome would provide abundant resources of candidate genes for economic traits and enable characterizing the functionality of the genes (Elsik et al., 2009; Zimin et al., 2009; Lee et al., 2013; K├Ąks et al., 2014).
In this study, we carried out whole-genome sequencing of Hanwoo bulls using Illumina Genome Analyzer IIx platform to discover SNP variation that may be related to economic traits and to characterize the genetics and molecular biology of Hanwoo, which would enable efficient Hanwoo breeding.
MATERIALS AND METHODS
Sample and DNA extraction
Ten Hanwoo bull semen straws were obtained from the Hanwoo Improvement Center of the National Agricultural Cooperative Federation, Seosan, South Korea, to represent Hanwoo breed. Many Hanwoo calves were born between the late 1980s and 2000 from the ten sires. The ten samples were chosen, so as not to be genetically related, and to reflect genetic diversity of the Hanwoo population. The genomic DNA was extracted using standard phenol/chloroform extraction method.
Sequencing, assembly and annotation
A genomic DNA library was constructed using a paired-end DNA sample prep kit following the manufacturerŌĆÖs protocols (Illumina Inc., San Diego, CA, USA). Purity and yield were confirmed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Sequencing results were analyzed using IncoAnalysis service at Insilicogen, Inc., YongIn, South Korea. For assembly and annotation of the sequence reads, two bovine genome assemblies named UMD3.1 (Zimin et al., 2009) and Btau4.6.1 (Elsik et al., 2009) were used as alternate reference sources. Using the CLC Assembly Cell Software 4.0.6, the pooled reads were aligned separately to the Btau 4.6.1 and UMD 3.1 using the parameters set as: Mismatch cost = 2; gap cost = 3; deletion cost = 3; high-scoring seqment pairs (HSP) coverage (length fraction) = 100%; Identity (similarity) = 98%. Sequence data were deposited in the NCBI database. Genomic (pseudo-molecules) sequences representing all Hanwoo chromosomes were constructed and annotated with corresponding bovine reference genomes, separately. The results were blasted against the UniProtKB database for gene ontology (GO) annotation. Each resultant pool-specific assembly was used to find SNP allele frequency differences and to calculate accumulated distribution per 10-kb interval on each chromosome.
SNP discovery
For SNP identification, sequence reads were mapped to the two bovine genome assemblies, Btau 4.6.1. and UMD 3.1, using the ŌĆśfind_variationsŌĆÖ program in CLC Assembly Cell along with stringent parameters as follows: minimum depth = 5; minimum mismatch count (show information when more than a given number of reads is different from the consensus) = 3; limit fraction (Show information when more than a given fraction of reads is different from the consensus) Ōēź20%. In order to compare our findings with dbSNP database, we carried out the liftover from dbSNP set to Btau 4.6.1 and UMD 3.1, respectively, since the dbSNP set is not yet mapped to the Btau 4.6.1 and UMD 3.1 assemblies. We thus compared our SNPs with dbSNPs (Btau 4.6.1 and UMD 3.1) mapped by SNP location of dbSNP. Heterozygous and homozygous SNPs were determined with 50% cut-off of aligned reads identifying the SNP, but homozygous alleles which be constituted exclusively same bases were applied in order to unravel Hanwoo-specific SNPs. The study for promoter and downstream SNPs was conducted in upstream 2 kb and downstream 1 kb of each gene, respectively.
Ontology analysis of SNP-containing genes
Ontology analysis was conducted SNP-containing genes through GeneCodis3 programme (http://genecodis.cnb.csic.es/). This analysis carried out based on GO, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, interpro motifs, and Panther pathways.
Genes related to economic traits
Five traits were focused to characterize genetic variants in the Hanwoo genome; meat quality including juiciness, tenderness, marbling (or IMF), milk and yellow hair. Pathways/groups were collected with entities and relations extracted from searching PubMed database together with MedScan reader v5.0 based on literature-search algorithm. The results were imported into Pathway Studio 9 (Elsevier Research Solutions, Amsterdam, Netherlands), in which pathway-based analysis was performed using mammalian ResNet database and customized bovine ResNet database. Conversion of genes having human Gene ID into bovine Gene ID was performed using HomoloGene, which is a database to automatically detect homologs including orthologs among the annotated genes of 20 completely sequenced eukaryotic genomes.
RESULTS AND DISCUSSION
Sequencing, assembly and mapping
Sequencing reads from the ten samples were pooled to perform the whole-genome sequencing analysis followed by SNP discovery. Sequencing generated 27 Gbp of high quality sequences on paired-end libraries (76-bp reads and 84-bp reads) with insert size of 500 bp. The genome sequence traces were submitted to the NCBI Short Read Archive (SRA) as accession SRP060306. Preprocessed clean reads were mapped on two reference genome assemblies, giving an average depth of 9.64-fold (UMD 3.1) and 8.58-fold (Btau 4.6.1) coverage, resulting in mapping of 97.20% and 91.97% to the reference genomes, UMD 3.1 and Btau 4.6.1, respectively (Table 1). Overall, the number of reads mapped to each bovine chromosome (Chr) was unbiased, except for Chr1, Chr8, Chr27, and Chr29 in the UMD 3.1 reference genome (Figure 1). The coverage of the mapped reads was fairly even on individual chromosomes (Supplementary Figure 1). This result is consistent with the results of whole-genome resequencing of Japanese native cattle Kuchinoshima-Ushi (Kawahara-Miki et al., 2011).
In the previous study, whole-genome sequencing revealed 98% next generation sequencing (NGS) reads (at 15.8-fold read depth) of Fleckvieh genome and 93% (at 7.4-fold read depth) of Kuchinoshima-Ushi genome were mapped to the NCBI bovine genome (Btau 4.6.1). The relatively lower genome coverage in this study may be caused by two reasons. Firstly, although mapping with default parameter (identity 80% and HSP coverage 50%) increased mapping coverage of 98.73% (UMD 3.1) and 93.79% (Btau 4.6.1), we produced reliable sequencing data with the optimized parameter, which caused the diminished coverage. Secondly, compared to Fleckvieh breed and Kuchinoshima-Ushi breed, Hanwoo is evolutionally more distant to the breed of the reference genome (Hereford). The different reference genome is attributable to different coverage, because Hanwoo genome had more similarity with the UMD 3.1 reference genome than the Btau 4.1.6 genome. Thus, the UMD 3.1 version was used for further analysis of Hanwoo genome. Substantially, UMD 3.1 and Btau 4.1.6 genome assemblies are significantly different in terms of several aspects (Kawahara-Miki et al., 2011). Direct alignment of two genomes against each other showed diverse discrepancies in ten chromosomes and most remarkable differences were observed in X chromosome (Chr X), though Btau was homologous to the human Chr X (Zimin et al., 2009). For another parameter of assembly completeness, the number of full-length cow mRNA mapping to the two reference genomes was different in accordance with levels of coverage, which reveals that UMD contains more mRNAs than Btau (Kawahara-Miki et al., 2011). Therefore, our suggestion is critical because, obviously, the mapped reads generated by the reference assembly method can vary depending on different reference genomes. Approximately 2.80% and 8.03% of the Hanwoo sequence reads were unmapped to each reference assembly (UMD 3.1 and Btau 4.6.1), possibly due to complexity of the breed or individual uniqueness, sequencing errors, or the current incompleteness of the Hanwoo sequencing.
High-quality SNP detection and novel SNPs
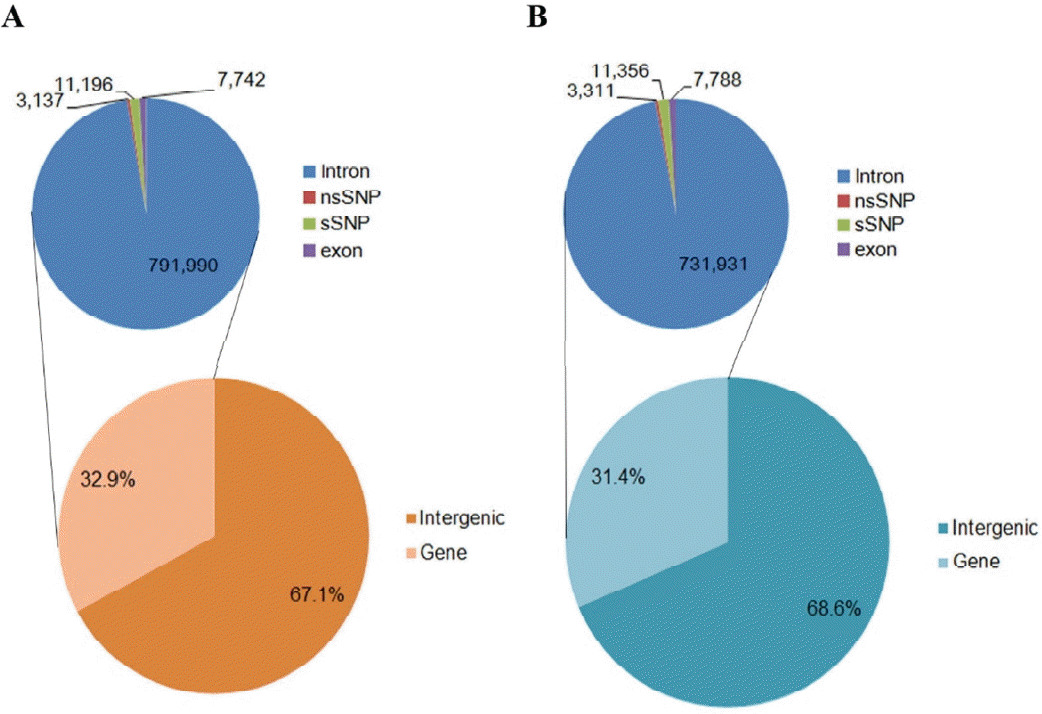
In order to reduce the false-discovery rate, stringent criteria for SNP calling were applied (materials and methods). As a result, 2,473,884 (UMD 3.1) and 2,402,997 (Btau 4.6.1) SNPs were identified. Among the SNPs, 2,234,514 (90.3%) were heterozygous and 239,370 (9.7%) were homozygous for UMD 3.1 (cut-off: 50% alleles), whereas 2,162,910 (90.0%) were heterozygous and 240,087 (10.0%) were homozygous for Btau 4.1.6 (Table 2). This result suggests that substantial variations exist between the ten individuals. Especially, 791,990 (32.9%) and 731,931 (31.4%) SNPs that were localized in genic regions. Synonymous SNPs (sSNPs) and nonsynonymous SNPs (nsSNPs) in exon region was 11,196 (1.38%) and 3,137 (0.39%) for UMD 3.1, while 11,356 (1.50%) and 3,311 (0.44%) for Btau 4.6.1 were defined in the exon regions for the sSNPs and nsSNPs, respectively (Figure 2). To discover novel SNPs, all the SNPs were compared to the locations where SNPS were already known from NCBI dbSNP database (Supplementary Figure 2). Amongst, 1,095,922 (44.3%) and 982,674 (40.9%) SNPs were novel for UMD 3.1 and Btau 4.1.6, respectively, (Table 2). Considering SNP density, 927 SNPs for UMD 3.1 and 809 SNPs for Btau 4.6.1 were discovered per 1 Mb.
Hanwoo-specific SNPs
Hanwoo-specific SNPs were determined by comparison of genomic position of each SNP with those of NCBI dbSNPs. We isolated 100% homozygous SNPs for discovering SNPs associated with Hanwoo-specific phenotypes. As a result, 46,301 SNPs (1.9%) for UMD 3.1 and 28,613 SNPs (1.2%) for Btau 4.6.1 were identified, respectively, (Table 3). Among the SNPs, 46,301 Hanwoo-specific SNPs that were based on UMD 3.1 were chosen for further analysis.
Functional classification of SNP-containing genes
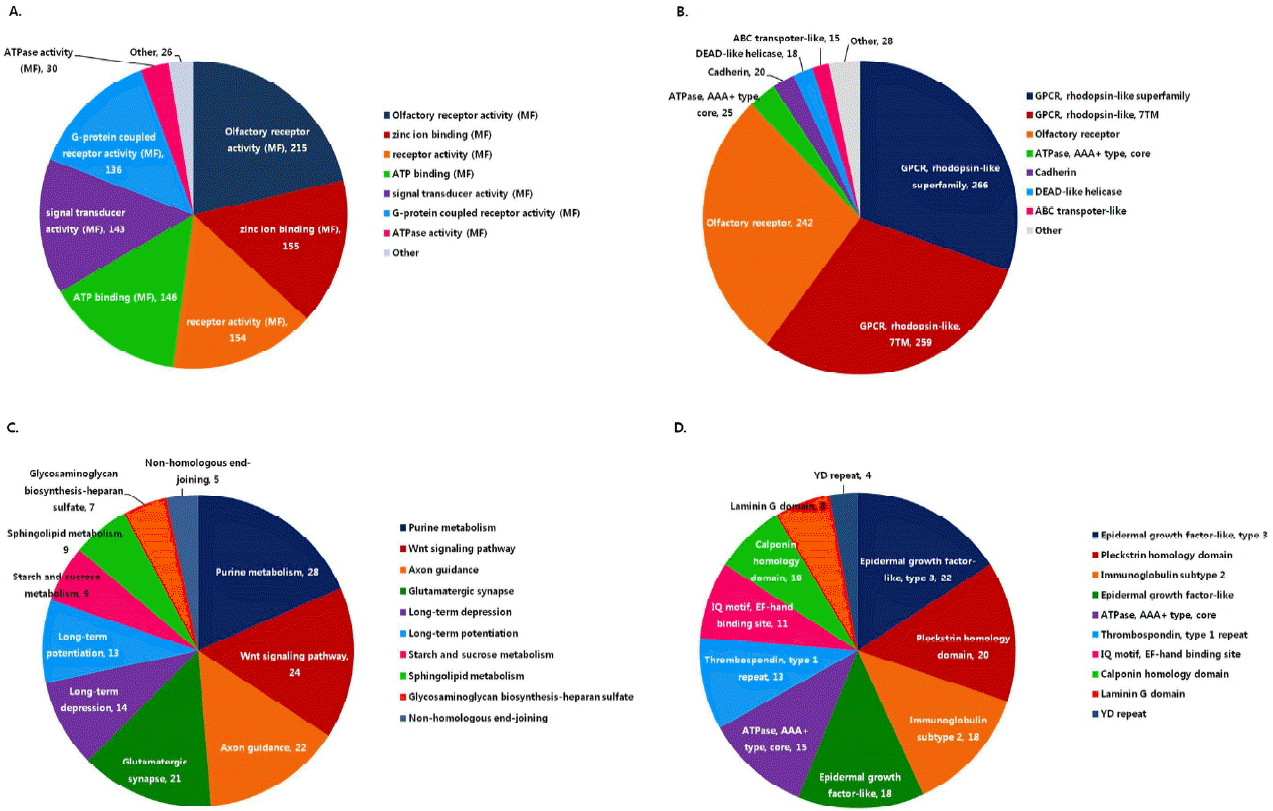
Non-synonymous SNPs (nsSNPs) may lead to phenotype change. To elucidate the effects of nsSNPs in Hanwoo, GO analysis was conducted in nsSNP-containing genes. A set of 2,329 nsSNP-containing genes was significantly enriched in GO including olfactory receptor activity (molecular function), zinc ion binding, G protein-coupled recepters (GPCR), rhodopsin-like superfamily (Figure 3). Majority of these genes are involved in signal transduction pathways that transfer the signals from outside of a cell through cell surface receptors to the inside of the cell including hormone and neurotransmitter. A similar result was observed not only in cow but also in human, implying that the genetic variation in signal transduction genes may be one of main causes of individual phenotypic diversity (Nusse and Varmus, 2012).
Hanwoo-specific SNP containing genes
A total of 5,022 genes were characterized as Hanwoo-specific SNP containing genes. Those genes were enriched in purine metabolism, Wnt signaling pathway, axon guidance, and glutamatergic synapse pathway (Figure 3). Axon guidance pathway represents a key stage in the formation of neuronal network, and glutamatergic synapse pathway is also the major excitatory neurotransmission in the mammalian central nervous system. These neuronal pathways can modulate immune-related system by releasing neurotransmitter (Kim et al., 2009). Especially, 581 genes that have Hanwoo-specific SNPs at least five are associated with immune activation by external stimuli, through epidermal growth factor-like domain (EGF-like domain), pleckstrin homology domain (PH domain), immunoglobulin subtype 2, and thrombospondin (Figure 3) (Chen et al., 2015). The Hanwoo-specific SNP containing genes might contribute to diversity of immune system.
SNPs-containing genes for economic traits
We collected the genes that are putatively associated with five traits of economic importance, i.e. milk production, marbling (or IMF), juiciness, tenderness, and yellow hair, by using MedScan Reader v5.0 (Ariadne Genomics, USA). The MedScan Reader is a natural processing language engine with text-mining algorithm that automatically extracts and displays information of interest from the biomedical literature in the PubMed and Google Scholar database. Using PubMed and Google Scholar server, we collected the research articles related with the traits of interest using relevant keywords. The information of entities and relations was extracted from the selected papers, but entities having no relations were used for this analysis. To obtain the accurate results, we checked whether the genes were involved in physiological processes associated with the five traits (Supplementary Tables 1 and Table 2).
SNPs-containing genes for yellow hair follicles
The pigmentation in mammals is based on the presence or absence of melanin in hair or skin. There are two types of pigments, i.e. eumelanins and pheomelanins which are responsible for black/brown and yellow/red colors, respectively. The major genes affecting the synthesis of these pigments are tyrosinase-related protein 1 (TYRP1), dopachrometautomerase (DCT), melanocortin receptor 1 (MC1R), tyrosinase (TYP), agouti signaling protein (ASIP) and corin, serine peptidase (CORIN). Especially, in the rodents, the change between black and yellow pigment is arbitrated by the interaction between MC1R and its antagonist ASIP. We analyzed the genetic variations of the genes associated with hair color traits including above genes using MedScan reader. It has been revealed that the T-to-C substitution in MC1R gene caused amino acid change in position 99, Leu to Pro in Angus black cattle. This shift allowed the expression of the constitutive activation of MC1R receptor, leading to a decrease in the Agouti concentration, and finally to determine the black color in Angus. Whereas, in the absence of Corin, a negative regulator of Agouti, Agouti activity is prolonged and determines the yellow color (Van den Bossche et al., 2012). Hanwoo has yellow hair follicles over the whole body, where no SNP in MC1R was observed, a set of 227 SNPs were identified by including the nsSNP in CORIN. The nsSNP (NW_003103974:4359766C>T) of CORIN leads to change of amino acid Glu (negative charge) >Lys (positive charge) in position 826. In addition, six SNPs were found in upstream promoter region of the gene, implying that those variations may affect CORIN gene expression. In previous studies, variations in MC1R and microphthalmia-associated transcription factor (MITF) were reported to be related with cattle follicles. However, the polymorphism of CORIN in Hanwoo was reported for the first time in this study. MITF, which is a master regulator of melanocyte development, is known to regulate expression of TYP, TYRP1, and DCT, by direct binding to cis-elements in their promoter sequences. MITF mutations associated with a dominant white phenotype were not found in Hanwoo. In other cattle breeds, there are many SNPs reported in color related genes such as TYP, TYRP1, ASIP, DCT, melanophilin (MLPH), c-Kit (KIT), c-Kit ligand (KITLG), which caused phenotypic variations in hair follicles (Van den Bossche et al., 2012). Also, many nsSNPs were found in those genes in Hanwoo. However, it is not likely that the SNPs cause significant changes in protein structure enabling it to cause Hanwoo hair coloring (Table 4).
SNPs-containing genes for beef palatability
Tenderness, juiciness, and marbling are key components of beef palatability. A set of genes were determined for tenderness (94 genes) and juiciness (121 genes) (Supplementary Table 2). Many SNPs from our study were previously reported in other cattle breeds (Table 4). For example, Leptin (LEP) that is related with tenderness contains one nsSNP (Cys>Arg) of 269 SNPs. Leptin receptor (LEPR) genes were associated with fat content in carcass meat in Nellore, Holstein-Friesian, Angus, Charolais, Hereford, and Simmental (Pinto et al., 2011). In this study, many nsSNPs were also observed in LEP and LEPR genes in Hanwoo (Table 4). Based on a mutation similar to the mKIAA0657 protein (titin, TTN), we hypothesized that the pressure of the strong selection for high marbling in Japanese Black breed increases the frequencies of the T, among T and A alleles at the TTN g.231054C > T, RPL27A g.3109537C > T and ankirin 2 genes c.*188G > A SNPs, respectively (Watanabe et al., 2011). Many nsSNPs were also found in meat quality related genes such as ankyrin 1 (ANK1), stearoyl-CoA desaturase (SCD), microtubule-associated protein 1B (MAP1B), NCK-associated protein 1-like (NCKAP1L), insulin-like growth factor binding protein 5 (IGFBP5) and mitochondrial poly (A) polymerase (MTPAP) (Table 4). Some of the SNPs were localized in promoter region, which can affect gene expression of the genes (Supplementary Table 2). Calcium activated neural protease (CAPN) on BTA29 containing a quantitative trait locus for meat tenderness activated by increased muscle calcium is responsible for meat tenderness through proteolysis. Calpastatin (CAST) is an inhibitory protein of CAPN and its inhibitory activity is an indicator of tenderness (Carnagey et al., 2008). The calpain-calpastatin enzyme complex regulates the rate of protein degradation for tenderness (Cafe et al., 2010). In our study, a total of 3,935 and 5,144 SNPs, including 58 and 82 Hanwoo-specific SNPs, were found in UMD and Btau, respectively (Supplementary Table 2). Particularly, the Hanwoo-specific SNPs will be a valuable genetic resource to characterize beef quality of Hanwoo.
SNPs-containing genes for marbling
Marbling (or IMF) is a multi-factorial trait, i.e. determined by multiple quantitative trait loci (QTLs), interacted with environmental factors. Marbling is one of the important indicators for palatability of beef (Lim et al., 2011). Certainly, IMF accumulation is attributable to multiple pathways regulating the balance between uptake, synthesis and degradation of triglycerides, rather than the involvement of a single pathway (Canovas et al., 2010). Marbling can be redistributed from other fat deposits including subcutaneous fat and visceral fat, which gave rise to inefficient meat production. Thus, genomic studies on marbling have been performed to discover marbling-specific genes including ribosomal protein L27a (RPL27A), sphingosine-1-phosphate receptor 1 (S1PR1), ADAM metallopeptidase with thrombospondin type 1 motif 4 (ADAMTS4), hormone-sensitive lipase (LIPE), peroxisome proliferator-activated receptor alpha (PPAR), CCAAT/enhancer binding protein (C/EBP), alpha (CEBPA), runt-related transcription factor 1 (RUNX1), and insulin (INS) in animal breeding researches (Lee et al., 2008). In this study, LEP, calpain 1, (mu/I) large subunit (CAPN1), CAST, adiponectin,C1Q and collagen domain containing (ADIPOQ), peroxisome proliferator-activated receptor gamma (PPARG), insulin-like growth factor 1 (somatomedin C) (IGF1), fibronectin ED-B+ region (FN1), sorbin and SH3 domain containing 1 (SORBS1), myosin, light chain 12A, regulatory, non-sarcomeric (MYL12B), SH3-domain kinase binding protein 1 (SH3KBP1), acyl-CoA synthetase long-chain family member 1 (ACSL1), myosin light chain kinase (MYLK), ATP-binding cassette, sub-family A, member 1 (ABCA1), insulin receptor (INSR), protein kinase C, alpha (PRKCA) and similar to Retinoblastoma-associated protein (RB1) were newly identified as marbling-related (Supplementary Table 2). As an inhibitor to the growth of muscle tissue, myostatin is involved in muscle marbling during fattening period by inhibiting myogenesis and promoting adipogenesis (Shibata et al., 2006). A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS4), which takes part in connective tissue degradation, is expected to be involved in a pivotal biological pathway for building up marbling. Also, as regulators of ADAMTS4, IL-17A, TNF╬▒, and TGF╬▓1 were connected with marbling. Peroxisome proliferator-activated receptor ╬│ (PPAR╬│), C/EBP╬▒, and RUNX1 were significantly and positively related to protein increase in intramuscular fat content (Lee et al., 2010). In this study, we identified a set of 232 genes and found 22 nsSNP in hemicentin 1 (HMCN1, 437 sSNPs, 3 nsSNPs) and EGF (124 sSNPs, 2 nsSNPs). Also, significant SNPs were found in the promoter region of RUNX1, ME1, and ASAP1. A previous study revealed that RUNX1 was significantly up-regulated as intramuscular fat content increased (Bagnato et al., 2008). Mutations in ME1 and ASAP1 gene were associated with meat quality traits in Angus and Nelore, respectively (Lee et al., 2013). Then Hanwoo-specific SNPs in above genes are a valuable genetic source to improve meat quality (Table 4).
SNPs-containing genes for milk production
A set of 202 genes were found for milk production (Supplementary Table 2). Hanwoo is known to have low milking ability, and there are few studies about genes for milk production in Hanwoo. A list of genes for milking metablism are growth hormone (GH), such as GH1 (1 sSNP), IGF1 (39 sSNPs), and IGF2 (2 sSNPs), prolactin (PRL, 3 sSNPs), LARGE like-glycosyltransferase (687 sSNPs), diglyceride acyltransferase (DGAT1, 2 sSNPs), alpha-lactalbumin (LALBA, 4 sSNPs), lactoferrin (LTF, 53 sSNPs) and signal transducer and activator of transcription 5A (STAT5A, 13sSNPs) (Supplementary Table 2). GH1 has significant positive effect on milk yield throughout lactation (Chanchai et al., 2010). The short isoform of prolactin (PRL) receptor has a suppressive role in PRL signaling to production of milk protein (Berlanga et al., 1997). The DGAT1 gene, involved in triglyceride synthesis, is suggested to be the causative gene for the QTL affecting milk fat on bovine chromosome 14 (Ron et al., 2007). The LALBA is necessary for lactose synthesis, which is important for milk production as an osmotic regulator of milk secretion. The STAT5A gene, as a member of the interferon-tau, plays major role in milk production (Khatib et al., 2008). Although all of these genes are known to be associated with milk production, the genes with sSNPs are required for additional investigation within diverse genomic sources encompassing several cattle breeds. Moreover, many nsSNPs were found in a set of 26 genes including FAT tumor suppressor homolog 3 (FAT3, 903 SNPs), family with sequence similarity 13, member A (FAM13A, 94 SNPs), ATP-binding cassette, subfamily G (ABCG2, 92 SNPs) and SREBP cleavage activating protein (SCAP, 69 SNPs), in which the relationship of the genes with milk production was not previously reported (Supplementary Table 2).
CONCLUSIONS
In this study, we investigated the genetic variations related with economically important traits by using whole-genome resequencing of Korean native cattle, Hanwoo. A total of 2.47 million SNPs were identified, of which more than 1.1 million SNPs (44%) were found to be novel. Among the SNPs, many of the nsSNPs were in the genes that were reported as candidates for QTL affecting economically important traits such as meat quality, milk production, and exterior traits in other cattle breeds. Since Hanwoo has been selected for growth, disease resistance and beef quality for the last several decades, there should be genetic variations between Hanwoo individuals, e.g. SNPs in the genes that are functionally related to the respectively traits, which was supported in this study. In particular, a set of 136 Hanwoo-specific genes including 446 SNPs were found in the genes related to the above traits. Among the genes, CAPN1, CAST, LEP, ANK1, ADIPOQ, IGF1, NR3C1, PPARG, and FN1 were included, which were known to be related with marbling, tenderness and juiciness. We identified Hanwoo specific genes, for yellow hair, meat quality, and milk production. Our results will provide valuable genetic resources for genome study of other cattle breeds as well as Hanwoo, and will facilitate the characterization of the genetic architecture of economically important traits in cattle.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







