



INTRODUCTION
Pork meat constitutes more than 40% of worldwide meat production, forming a prominent source of human food (Rothschild et al., 2011). Customer demand for pork depends upon the meat quality and its physical and biochemical components (Bonneau et al., 2010). Thus, pork quality is an economically important factor and one of the major selection benchmarks for breeding process in the swine industry (Luo et al., 2012). With the advancements in meat processing technologies, it is now possible to predict ham, loin primal, and sub-primal cut weights (van Wijk et al., 2005). These developments paved the way for the pork industry to adopt more precise value-based classifying systems to satisfy the demands of pork market (Brorsen et al., 1998).
Pig breeding programs have been implemented mainly towards the reduction of production cost in the last several decades. Selection was, therefore, focused on economically important traits such as litter size, weight gain, back fat, and feed conversion. Recently, breeding objectives have also put weight on retail carcass yield and meat quality (van Wijk et al., 2005). Carcass quality traits are highly heritable, causing an efficient selection response (Newcom et al., 2002).
The Berkshire breed (Sus scrofa domesticus) was found 300 years ago in the Berkshire county of United Kingdom. The king of England preferred Berkshire pork for his own personal meat supply, because of excellent meat quality (American Berkshire Association, 2013). They have a dark skin, which protects them from sunburn. Some parts of body such as legs, face and tail, are white pointed. Berkshire pigs are characterized with pink skin color and a strong body type with short neck. Legs of the breed are short and blocky and feet are strong. An adult pig of the breed has an average weight of 272 kg (600 pounds). Individuals of the breed are usually friendly and curious and exhibit excellent disposition (Kawaida, 1993).
Recently, the demand for Berkshire pork has increased in Asia (McLaughlin, 2004), because of excellent meat quality such as richness, texture, marbling, juiciness, tenderness and flavor (Goodwin and Burroughs, 1995; Brewer et al., 2002). The thin muscle fibers and excellent water holding capacity (WHC) of the meat increases its popularity (Goodwin and Burroughs, 1995). The Berkshire pork is exported mainly to Japan and US (Lammers et al., 2011).
The evaluation of breeding values based on pedigree information has many limitations (Dekkers et al., 2010). Some phenotypic traits are difficult and costly to measure, resulting in low accuracy of estimated breeding values (Badke et al., 2014). These shortcomings could be overcome by using a genome based best linear unbiased prediction method (GBLUP), in which genomic estimated breeding values (GEBV) were predicted with a high density marker map covering the porcine whole genome (Meuwissen et al., 2001; Dekkers et al., 2010).
Genomic selection is a selection decision made at an early stage based on GEBV (Hayes et al., 2009). The main benefits of genomic selection are genetic enhancement by minimizing the generation interval and yielding a higher accuracy of estimated breeding values (Hayes et al., 2009; van Raden et al., 2009; Christensen et al., 2010; Wiggans et al., 2011; Boddhireddy et al., 2014).
The GEBVs can be predicted with dense, genome-wide maps of single nucleotide polymorphisms (SNPs), which can lead to a more precise prediction of pig breeding value at a young age. Cleverland et al. (2010) reported that GEBV accuracies in pigs were as good as those in dairy cattle, if the training population size was large enough. Important factors on the accuracy of genomic predictions include number of phenotypes, (i.e. training data, used to form the prediction equation), heritability, effective population size, genome size, marker density, and genetic architecture of the trait, in particular number of loci affecting the trait and distribution of their effects (Daetwyler et al., 2008; Goddard, 2009; Meuwissen, 2009). Recently, Bayesian methods have gained popularity in evaluating genomic selection, due to the fact that different variance is fitted to each SNP (Fernando et al., 2007; Moser et al., 2009).
In this study, we evaluated accuracy of GEBV for port quality traits in a Berkshire population under the GBLUP and Bayes B models.
MATERIALS AND METHODS
Animals and phenotypes
A set of Berkshire samples (n = 1,205) were collected in Dasan breeding farm, Namwon, Cheonbuk province, Korea, between 2008 and 2013. The piglets were weaned at 3 to 4 weeks of age and moved into piglet pens, in each of which about 100 piglets were raised for 60 days. Then, the pigs were placed in growth/fattening pens 20 pigs in size for 90 to 120 days. The pigs were fed with the commercial feeds according to the regiments of Purina Ltd. The samples were slaughtered approximately 211 (┬▒23) days of age in an abattoir in Namwon and cooled at 0┬░C for 24 h in a chilling room. Among the carcasses, 801 and 404 samples were analyzed for meat quality and composition in the laboratories of National Livestock Research Institute in Suwon and Sunchon National University in Sunchon, Korea, respectively. A total of 16 carcass and meat quality traits were considered: 1) back fat thickness (BF), 2) Commission Internationale de lŌĆÖEclairage (CIE) a, 3) CIE b, 4) CIE l 5) collagen, 6) carcass weight (CWT), 7) drip loss, 8) fat, 9) heat (cooking) loss, 10) moisture, 11) National Pork Producers Council (NPCC) color score, 12) NPCC marbling score, 13) pH24, 14) protein, 15) shear force, and 16) WHC. For each individual, slaughter age (sage), gender, and year-season of birth were recorded.
The CWT and BF of each carcass were measured. The parts of loins (longissimus dorsi, LD) on the left side of the cold carcasses were used to determine meat quality parameters. As soon as all samples were placed in vacuum bags, the samples were transported to the laboratory and then frozen at ŌłÆ50┬░C until they were analyzed. The middle portions of each loin were used for experiment. For analysis of moisture content, fat content, drip loss, and heat loss, the only subcutaneous fat of meat samples was removed. For the others, all visible fat was trimmed off.
The proximate composition of each LD muscle was obtained with a slightly modified method of AOAC (2000). Briefly, moisture content was measured by drying 3 g of samples place in aluminum dishes at 104┬░C for 15 h. The crude protein contents were measured by the Kjeldahl method (VAPO45, Gerhardt Ltd., Idar-Oberstein, Germany). The crude fat contents were extracted according to the method described by Folch and Sloane-Stanley (1957). The total collagen content was determined by measuring hydroxyproline and using a multiplication factor of 7.14 (Etherington and Sims, 1981). The surface color and marbling scores of each loin was categorized based on NPPC standard (NPPC, 2000).
The surface color value were measured by the CIE L*, a* and b* system using a Minolta colorimeter (Model CR-410, Minolta Co. Ltd., Osaka, Japan). The colorimeter was calibrated against a white reference tile plate (L* = 89.2, a* = 0.921, b* = 0.783), and the diameter size of aperture was 4 cm. The color L* (lightness), a* (redness), and b* (yellowness) values were obtained after 30 min blooming at room temperature. The average value of five random measurements taken from different locations was used for the statistical analysis.
The pH value of each meat sample was determined with a pH meter (Orion 2 Star, Thermo scientific, Beverly, MA, USA). Water holding capacity was determined by centrifugation. Briefly, 5 g of minced meat sample was placed into a centrifuge tube with a filter paper (No. 4, Whatman International Ltd., Maidstone, England), and centrifuged at 3,000├Śg for 10 min. WHC was calculated as the remaining moisture in the meat sample on the basis of the moisture content of the original meat sample. The drip loss was measured as the percentage weight loss of a standardized (3├Ś3├Ś3 cm) meat sample placed in a sealed petri-dish at 4┬░C during the storage of 2 d. The heat (cooking) loss was determined as the percentage weight loss of a standardized (3├Ś3├Ś3 cm) meat sample after cooking in an electric grill with double pans (Nova EMG-533, 1,400 W, Evergreen enterprise, Seoul, Korea) for 90 s, until the internal temperature of the meat sample reached 72┬░C.
The samples were prepared in a cubic form (30├Ś30├Ś20 mm), heated until internal temperature of the samples reached 72┬░C┬▒2┬░C, and then cooled for 30 min at room temperature. Each sample was cut perpendicular to the longitudinal orientation of the muscle fiber with a Warner-Bratzler shear attachment on a texture analyzer (TA-XT2, Stable Micro System Ltd., Surrey, UK). The maximum shear force value (kg) was recorded for each sample. Test and pre-test speeds were set at 2.0 mm/s. Post-test speeds were set at 5.0 mm/s.
Molecular data
The 1,205 pigs were genotyped with the Illumina Porcine 62 k SNP chips, in which a total of 62,163 SNPs that covered the entire porcine genome were embedded. To evaluate GEBV, the SNPs on 18 autosomal chromosomes were considered for quality control tests. Any SNP was excluded with <90% call rates, <5% minor allele frequency, or significant departure from Hardy Weinberg equilibrium (p<0.001). Those individuals with less than 90% genotyping call rate were also removed. After the quality control procedures using PLINK v7.0 (Purcell et al., 2007), 36,605 SNPs for 1,191 individuals were used. An imputation procedure was applied to predict missing genotypes of the SNPs with Beagle vs3.3.2 (Browning and Browning, 2007; Nothnagel et al., 2009).
Statistical analysis
To obtain GEBV under the GBLUP model, a linear mixed (Animal) model was fitted with the fixed effects of gender for pH24 and BF, and birth year-season for CWT, pH24, CIE a, CIE b, CIE L, drip loss, heat loss and shear force, and a covariate, slaughter age for fat, protein and BF, respectively. Statistical significance of the fixed factors or covariate was tested using SAS general linear model procedure (vs9.2). For the rest of meat quality traits, the effects of the factors were not significant (p>0.05). The Animal model, then, can be written as
where y is the vector of phenotypic record of the animal, b is the vector of overall mean, fixed and covariate effects, g is the vector of breeding values, X is the design matrix for the fixed and covariate effects, Z is the design matrix allocating records to breeding values and e is the vector of the residual of the phenotype. To construct genome relationship matrix (G), the subroutine in R was used (version 2.15.0), which was then incorporated into the Animal model using ASREML (average sparsity residual maximum likelihood) 3.0 (Gilmour et al., 1995). The mixed model equation was then
where ╬▒ = Žāe2/Žāg2 = (1ŌĆōh2)/h2, Žāe2 is the residual variance, Žāg2 the genetic variance, and h2 heritability.
The breeding values for both phenotyped and non-phenotyped individuals can be predicted by solving the equation:
G matrix was calculated based on the observed allele frequencies of the markers. The equation used to calculate G matrix was:
The marker matrix, M, had order of n├Śm, in which n is the number of individuals and m is the number of markers. In the M, alleles were coded as AA (homozygous for the first allele) = ŌłÆ1, AB (heterozygous) = 0, BB (homozygous for the second allele) = 1. The elements of P matrix were calculated using the formula Pj = 2(PjŌĆō0.5), where Pj was the minor allele frequency of the marker locus j. (M-P) is called the incidence matrix (Z) for markers. The P matrix was subtracted from the M matrix to set the mean values of the allele effects to 0, and to give more credit to rare alleles than to common alleles. Genomic inbreeding coefficient would be greater if the individual is homozygous for rare alleles than if homozygous for common alleles.
The meat quality traits composed different sets of training (with phenotype records) and testing (without phenotypes) data. For each individual, GEBV value was predicted, and the expected accuracy of GEBV for the ith individual was calculated using standard errors of GEBV as
The mean and standard deviation of the GEBV accuracies was calculated for each trait.
Evaluation of the GEBV was also carried out under the Bayes B model using Gensel 4.0 (Fernando and Garrick, 2008) software. The Bayes model was:
where
y = the vector of phenotypes
╬╝ = overall mean.
X = the incidence matrix of the fixed and covariate effects.
b = the vector of fixed and covariate effects.
zi = a vector of genotypes of a fitted marker i, that is coded as ŌłÆ10, 0, or 10.
ai = a random substitution effect of the fitted marker i with its variance, Žāai2.
e = the vector of random residuals that was assumed to be normally distributed.
For the marker effects, a mixture model was applied, i.e. a fraction of markers (ŽĆ) with zero effect and 1-ŽĆ of markers with non-zero effects, which was used to predict GEBV. Then, the genetic variances of the markers with non-zero effects would have Žāai2>0 (Habier et al., 2011).
The ŽĆ values ranged between 0.996 and 0.999 depending on the traits with different sizes of the training data.
The fixed effects for each trait were fitted as under the GBLUP model. The estimates of genetic and residual variances that were obtained from ASREML analysis were used as prior values for the Bayes B analysis. A total of 41,000 iterations of Markov chain were run for the analyses, with the first 1,000 iterations of burn-in period and each of 100 iterations was selected to calculate posterior mean and variance for the marker effects. The GEBVs were based on a weighted sum of the number of copies of the more frequent allele at each SNP locus, with the weights being the estimated allele substitution effects (╬▓). The sum of all the marker scores for an individual gave the genomic breeding value.
RESULTS AND DISCUSSION
Summary statistics for the sixteen meat quality traits were displayed in Table 1. The coefficient of variation were various between traits, e.g. 1.4% for moisture and 51% for drip loss. A set of 36,605 SNPs was chosen from the 62,163 SNPs in Illumina Porcine 60 k Beadchip (Table 2). The number of SNPs (4,426) was the greatest in Sus scrofa chromosome (SSC) 1, while SSC18 had the smallest number of SNPs (886). The SSCs 2, 4, 6, 7, 8, 9, 13, and 14 had more than 2,000 SNPs. The physical map with all of the available SNPs spanned about 2,195 Mb with an average distance of 67.9┬▒106.7 Kb between adjacent SNPs. However, the average distances were various between chromosomes, ranging between 51.9 Kb in SSC14 and 92.4 in SSC15.
The heritabilities that were estimated using genome relationship matrix (G) ranged between 6% and 46% (Table 3). Tomiyama et al. (2011) reported 0.54 and 0.32 for (BF) at finish and CWT, respectively, in a Japanese Berkshire population (n = 4,773). Jung et al. (2011) reported that heritabilities of pH2 4 h, CIE a, b, and L, WHC, NPPC marbling, drip loss, heat loss and shear force ranged between 0.51 and 0.66 in a Berkshire population (n = 808), Korea. Compared with the two reports, the heritability estimates of the meat quality traits in this study were low.
The GEBV accuracies of the testing as well as training data were calculated using the GBLUP and Bayes B methods (Table 3). Under the GBLUP model, the average (┬▒standard deviation) of GEBV accuracy means across the traits was 0.65┬▒0.04 for training data, which ranged from 0.42┬▒0.08 for collagen to 0.75┬▒0.02 for WHC. Under the Bayes B model, the GEBV accuracy mean ranged from 0.10┬▒0.14 for NPCC marbling score to 0.76┬▒0.04 for drip loss with the average of 0.49┬▒0.10 across all the traits. For the testing data sets, the GEBV accuracy was lower, i.e. the overall average of the traits was 0.46┬▒0.10 under the GBLUP model, ranging from 0.20┬▒0.18 for protein to 0.65┬▒0.04 for drip loss, and 0.38┬▒0.13 under the Bayes B model, ranging from 0.04┬▒0.09 for NPCC marbling score to 0.72┬▒0.05 for drip loss (Table 3).
For all the traits, the GEBV accuracies were greater for the training samples than for the testing samples under both the GBLUP and Bayes B models. This makes sense in that, for the training samples, the GEBV prediction was based on both genotypes and phenotypes, while only the genotype information was exploited for the testing samples. However, the GEBV accuracy differences between the training and the testing samples were smaller for the traits with great training sample size. For example, the average GEBV accuracy of CWT (moisture) under the GBLUP model, with 1,051 (693) training individuals, was 0.60 (0.74) for the training and 0.56 (0.54) for the testing samples, respectively (Table 3).
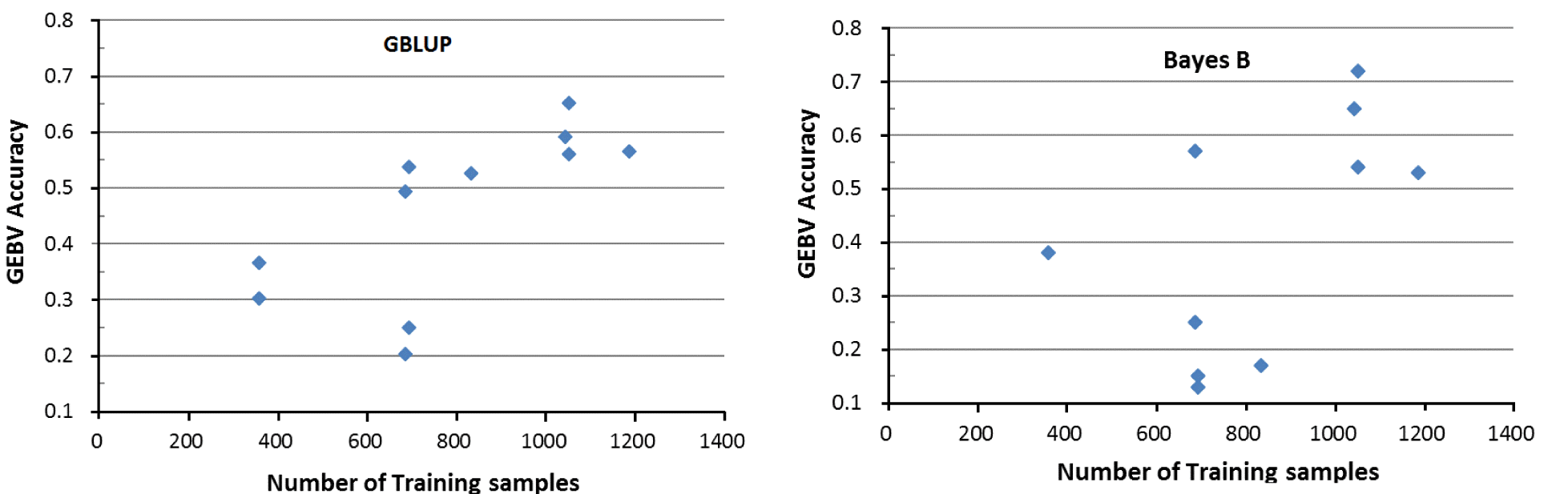
The GEBV accuracy depends on four factors; 1) size of the training population, 2) the heritability of each trait, 3) the extend of linkage disequilibrium between the markers and the QTL, and 4) the distribution of QTL effects (Goddard, 2009; Hayes et al., 2009). Our results supported the first two factors. There was a general tendency of high GEBV accuracy of testing samples with the training sample size (Table 3) under both the GBLUP and Bayes B models (Figure 1). For example, the average GEBV accuracy of NPCC color score (drip loss) for the testing samples, with 358 (1,051) training samples, was 0.30 (0.64) under the GBLUP model. For the traits with the sample training sample size (e.g. CWT and drip loss, n = 1,051), the testing samples had greater GEBV accuracy for drip loss (average was 0.72 under the Bayes B model) than for CWT (0.54), for which heritability of the former (latter) trait was 0.27 (0.13) (Table 3).
In general, the average GEBV accuracy values were similar between the GBLUP and Bayes B methods. However, for some traits with small sample size, e.g. collagen, moisture and NPCC marbling score, the accuracy of both under the Bayes B model was much smaller than under the GBLUP model (Table 3). This may be partly due to small sample size of the training data, which would cause the estimation of GEBV to be more sensitive to the prior values of the Bayes B method. Cleveland et al. (2010) reported that GEBV accuracy decreased with small training sample size, because of not enough information to accurately estimate SNP effects.
There were a few reports about GEBV accuracy in pig populations. Badke et al. (2014) reported that the GEBV accuracy for BF was 0.45 to 0.47 in a Yorkshire population (965 training samples). Our results showed that, for the trait, 0.59 and 0.65 accuracies were obtained by the GBLUP and Bayes B analyses, respectively, using 1,043 training samples (Table 3).
There are many reports about successful genomic selection procedures in dairy cattle (Hayes et al., 2009; van Raden et al., 2009; Wiggans et al., 2011). The dairy cattle breeding industry is benefitting from genomic selection mainly by reduced generation intervals (Wellmann et al., 2013), while, in the pig industry, the benefit from genome selection was less, partly due to short generation intervals (Wellmann et al., 2013). However, some studies reported that genomic selection was relevant in pig breeding, by improving maternal traits (Simianer, 2009; Lillehammer et al., 2011) or by boosting selection intensities (Tribout et al., 2012; Wellmann et al., 2013).
CONCLUSION
Herein, we presented the first report about GEBV accuracy of a Berkshire population in Korea, and our results were in general agreement with the previous GEBV studies, i.e. GEBV accuracy depends on the size of training data as well as heritability of the tested trait. For some traits such as drip loss, GEBVs were predicted with a good accuracy under the GBLUP or Bayes B model (Table 3). However, more training samples are needed to further improve the GEBV accuracy for pork quality, especially with low heritable traits, to efficiently implement genome selection programs in Berkshire industry in Korea.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







