





INTRODUCTION
The discovery and development of a new drug can save millions of lives and enhance the quality of numerous lives. Although the pharmaceutical and biotechnical researches and developmental efforts may lead to successful approval, there are also many hurdles in drug discovery and development [1]. Most of all, in order to obtain regulatory approval, we have to discover a drug candidate which shows proper efficacy, safety, and drug-like properties [2,3]. A good strategy for achieving successful clinical trials is to determine absorption, distribution, metabolism, excretion, and pharmacokinetics of a drug including physicochemical properties such as LogP and solubility [4,5].
Activation of the interleukin-6 (IL-6) and signal transducer and activator of transcription factor-3 (STAT3) signaling pathway have been studied and IL-6, a phosphorylated glycoprotein, is a major causative factor which plays an important role in inflammatory disease [6,7]. IL-6 plays an important role in anti-inflammatory and host defense and overexpression of IL-6 due to pathogens, stresses, and aging may lead to the pathogenesis of inflammatory processes, autoimmune response, and cancer [8–10]. The therapeutic antibody (Ab) agents which inhibit IL-6 have been developed and showed effective and potential alternatives for inflammatory diseases refractory to conventional drugs. Recently, clinical studies have either been done or are ongoing on therapeutic antibodies such as sirukumab (CNTO136), siltuximab (CNTO328), olokizumab (CP6038), elsilimomab (BE-8), clazakizumab (BMS945429), PF-423691, and MED I5117, and by anti-IL-6R Abs including sarilumab (REGN88) and tocilizumab (Actemra) [11]. Even though these mAb biologics targeting IL-6 have been showing success in commercial view, they have several issues such as high cost, invasive administration, and high rate of immunogenicity. Therefore, the discovery and development of new oral absorptive small molecular inhibitors are required, particularly with the attributes of low toxicity and low antigenicity.
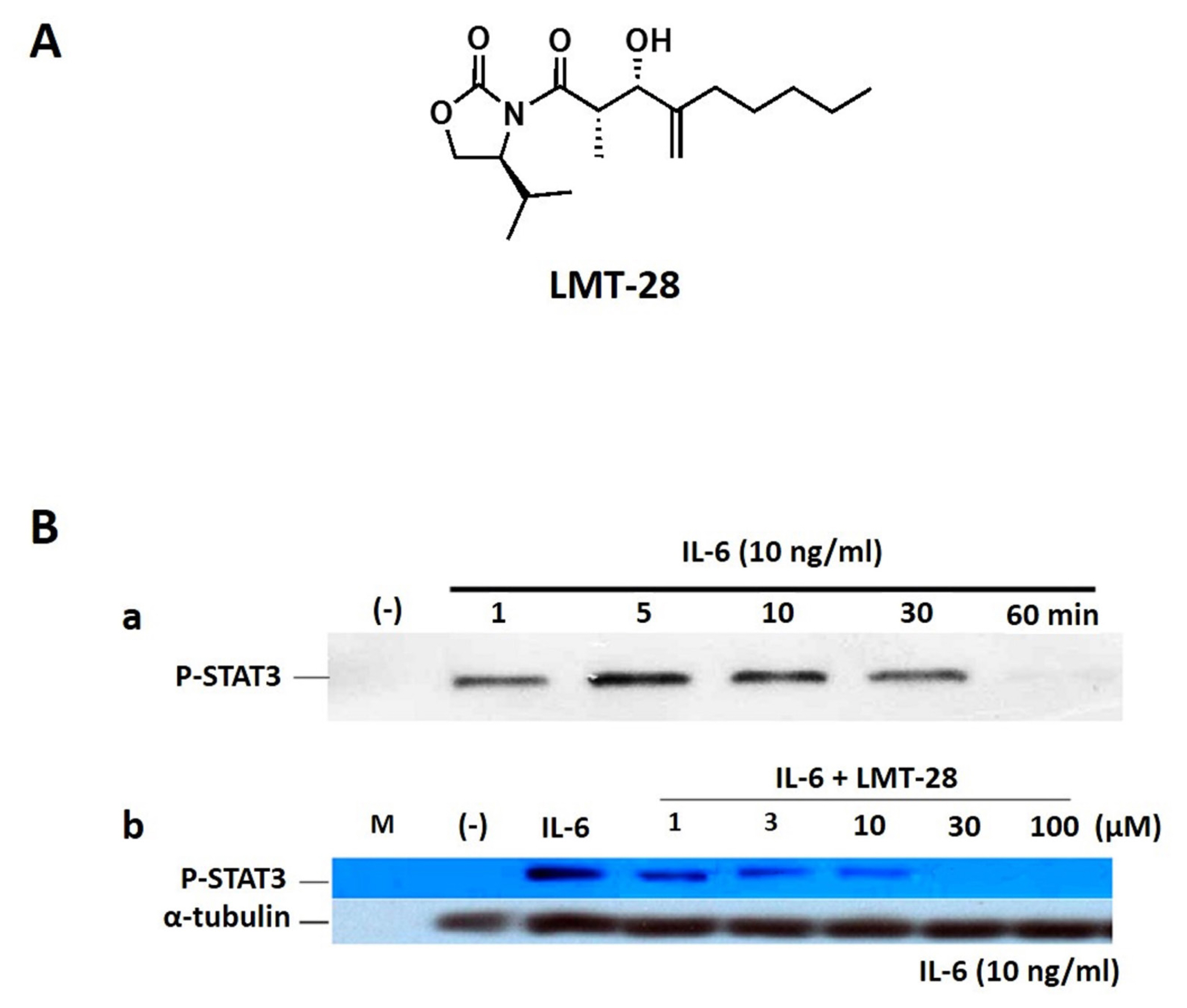
To discover a novel synthetic small molecule as a new drug candidate for IL-6 blocking, LMT-28 (Figure 1A) has been screened, identified from chemical library and investigated for its activity and mechanism of action [12–15]. LMT-28 has an inhibitory effect on the activation of STAT3 induced by IL-6. LMT-28 also has a down-regulatory effect for IL-6-stimulated phosphorylation of STAT3, gp130, and JAK2 protein. In addition, LMT-28 directly interacts with gp130 and specific reduction of IL-6/IL-6Rα complex binding to gp130. Taken together, it is reasonable that LMT-28 can be a first synthetic IL-6 inhibitor with IL-6 inhibitory activity and non-cytotoxicity.
The aim of this study was therefore to evaluate pharmaco kinetic profiles of LMT-28 as a novel IL-6 inhibitor, because there are no reports about characterizing it as a synthetic small molecule that inhibits IL-6 and ameliorates inflammation in mice by functioning through direct interaction with gp130. Furthermore, both in vitro and in vivo pharmacokinetic profiling including physicochemical properties were characterized to support future development and clinical trials.
MATERIALS AND METHODS
Chemicals
LMT-28 (Figure 1A, M.W. 311.42), an oxazolidinone derivative, was synthesized with a purity >99.0%. Liver microsome and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) regeneration solution for metabolic stability were purchased from Corning Gentest (Tewksbury, MA, USA). Carbamazepine as an internal standard along with other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Water, acetonitrile and other solvents high-performance liquid chromatography (HPLC) grade or the highest quality were purchased from J.T. Baker (Phillipsburg, NJ, USA).
Animals
Male BALB/c mice (CD-1) were purchased from Orient-Bio Company (Seongnam, Korea). Animals were kept in an air conditioned room at a temperature of 20°C to 25°C, humidity (50%±3%), and 12 h day/night cycles with specific free condition. Food and water were supplied ad libitum. Animals were fasted for 12 h before experiments while still having access to water. All animal procedures involving animal care were approved by the Institutional Animal Care and Use committee at Kangwon National University (approval no. KW-151127-2).
Western blot analysis for inhibitory effect of interleukin-6-induced signaling
HepG2 cells were seeded in MEM media (Welgene, Gyeongsan, Korea) with 10% (v/v) feta bovine serum, streptomycin (100 U/mL), and penicillin (100 U/mL; Life Technologies, Carlsbad, CA, USA). After seeding, cells were starved overnight, treated with LMT-28 for 1 h, and stimulated with IL-6 (10 ng/mL; R&D Systems, Minneapolis, MN, USA) for 10 min. Cells were treated with cell lysis buffer (Promega, Madison, WI, USA) for 30 min on ice. Cell lysates were separated on an 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, then transferred to nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). The membranes were blocked overnight in Tris-buffered solution (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% Tween 20) containing 5% nonfat dry milk at 4°C and then incubated for 1 h with the appropriate primary Abs against p-STAT3 (MilliporeSigma, Burlington, MA, USA) or α-tubulin in Tris-buffered solution (TBST; 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% Tween 20) containing 5% nonfat dry milk at room temperature. The membranes were washed (10 min, five times) and incubated for 1 h with horseradish peroxidase-conjugated secondary Abs diluted to 1:6,000. After 10 washes, the membranes were incubated with enhanced chemiluminescence reagents (MilliporeSigma, USA) and chemiluminescent signals were visualized using X-ray film.
Physicochemical properties of LMT-28
The shake-flask method was applied to measure the equilibrium solubility of LMT-28 [16,17]. Excess amount of LMT-28 was added into the water and the suspension was shaken at 25°C during 24 h in a flask for equilibrium solubility. The solution, then, was filtered and the concentration of LMT-28 in the filtered solution was quantified using ultra HPLC (ThermoFisher Scientific, Waltham, MA, USA) at wavelength 254 nm. The lipophilicity of LMT-28 was measured by using GLpKa (Pion, Cambridge, MA, USA) based on dip-probe absorption spectroscopy (D-PAS) method. The resultant values were calculated by LogP software (Pion, USA) and the computational predictions for number of hydrogen bond donors, hydrogen bond acceptors, rotatable bonds and predicted polar surface area (PSA) and LogS were made using Percepta software (ACD/Labs; Toronto, Canada).
Metabolic stability of LMT-28
Metabolic stability using rat and human liver microsomes (Corning Gentest, USA) of LMT-28 was performed according to previous reported protocol [18]. In brief, NADPH regeneration solution (Corning Gentest, USA) was added to initiate the metabolic reaction after mixing LMT-28 (final concentrations; 1 μM) with human or rat liver microsomes (0.5 mg protein/mL) in 100 mM potassium phosphate buffer (pH 7.4). After incubation at 37°C at 0, 10, 30, and 60 min, the reaction was terminated by adding three times of ice-cold acetonitrile containing internal standard (carbamazepine 10 ng/mL). After mixing by vortex and centrifugation at 10,000 g for 5 min at 4°C, the supernatants were transferred to fresh LC vials. Samples (5 μL) were analyzed by directly injection into a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system for the quantification of LMT-28.
The metabolic half-life and in vitro intrinsic clearance of LMT-28 were calculated by next equations [19].
where K is the elimination rate constant of LMT-28 in the microsomal metabolic reaction, half-life (t1/2) is microsomal metabolic half-life, CLint, in vitro is in vitro intrinsic clearance, and CLint, in vivo is in vivo intrinsic clearance. The liver weight relative to the body weight in the rat and human are 40 and 21 g/kg, respectively [20].
In vitro pharmacokinetic study of LMT-28
In order to determine cell permeability of LMT-28, Madin-Darby canine kidney (MDCK) cells were cultured and seeded at a density of 6×104cells/cm2 in 12-well Transwell plates. After checking cell monolayer integrity, the transepithelial electrical resistance (TEER) values (>500 ohms) were measured using an EVOM epithelial voltohmmeter (World Precision Instruments, Sarasota, FL, USA). After pre-incubation for 30 min at 37°C, LMT-28 (final concentrations of 2, 10, and 50 μM) was added to the donor wells in transport assay system. Sample aliquots (100 μL) were collected every 30 min for 120 min, and the same volume of fresh buffer added. The aliquots were stored at −20°C until quantitative analysis of LMT-28 concentrations by LC–MS/MS system. The apparent permeability coefficient (Papp, cm/s) was calculated as following equation:
where dQ/dt is the permeation rate across cell monolayer, A is the surface area of the monolayer (0.33 cm2), and C0 is the initial donor concentration.
In vivo pharmacokinetic study of LMT-28 in mice
To evaluate in vivo pharmacokinetics in mice, LMT-28 was dissolved in phosphate-buffered saline (pH 7.4) containing 5% (w/v) carboxymethyl cellulose (MilliporeSigma, USA) and administered orally by using oral needle and intravenously in the mouse tail vein at the dose of 5 mg/kg to male BALB/c mice (22 to 25 g) after overnight fasting. Blood samples (50 μL) were collected at 0.5, 1, 2, 4, 6, and 8 h after oral administration of LMT-28. After taking the blood samples from mice, the samples were centrifuged to obtain plasma samples at 4°C and 10,000 g and obtained plasma samples were stored at −20°C. These plasma samples (25 μL) were used for the quantification of LMT-28 concentration in mice.
Quantitative analysis of LMT-28 in biological fluids using liquid chromatography-tandem mass spectrometry
The concentrations of LMT-28 in plasma or microsomal reaction solution were determined by LC-MS/MS. For the sample preparation before LC-MS/MS analysis, three times (75 μL) of acetonitrile containing internal standard (carbamazepine 10 ng/mL) were added to 25 μL aliquots of the plasma samples. The samples were vortex-mixed and centrifuged at 10,000 g for 5 min at 4°C for the protein precipitation. The supernatant (90 μL) was transferred to LC vials and 5 μL was injected onto a LC-MS/MS system.
Mass spectrometric detection of LMT-28 was performed on an API 4000 Qtrap mass spectrometer (AB Sciex, Foster City, CA, USA) equipped with a positive electrospray ionization source. The chromatographic separation was performed on Hypersil Gold C18 column (50 mm×2.1 mm i.d., 3 μm particle size; ThermoFisher Scientific, USA) with a flow rate of 0.3 mL/min. The positive multiple reaction monitoring (MRM) mode were based on the most abundant product ions at m/z 312 → 165 for LMT-28, and m/z 237 → 194 for internal standard. Other parameters were the nebulizing (GS1), 50 psi; heating (GS2), 50 psi; collision energy, 23 eV for LMT-28 and 15 eV for internal standard, respectively. Analyst software version 1.4.2 (AB Sciex, USA) was used for the analysis of LMT-28 concentrations.
Pharmacokinetic and statistical analysis
The plasma concentrations versus time profiles of LMT-28 in mice were analyzed by a non-compartmental model using WinNonlin software ver 5.3 (Pharsight, Mountain View, CA, USA). The pharmacokinetic parameters such as area under the plasma concentration-time curve (AUC), peak plasma concentration (Cmax) and the time to reach a peak concentration (Tmax), terminal elimination half-life (t1/2), mean residence time (MRT) after oral administration were obtained from each mouse’s plasma concentration-time plots for LMT-28. All data were expressed as the mean±standard deviation. A value of p<0.05 by student t-test or analysis of variance analysis was considered to be statistically significant.
RESULTS
Inhibition of interleukin-6-induced signaling by LMT-28
As shown in Figure 1, the inhibitory effect of LMT-28 on IL-6-induced signaling was investigated. First, to obtain the ideal time condition for IL-6 treatment, HepG2 cells were treated with IL-6 at a concentration of 10 ng/mL for 1, 5, 10, 30, or 60 min and p-STAT3 was detected by Western blotting (Figure 1B). HepG2 cells which were pre-treated by LMT-28 (1, 3, 10, 30, and 100 μM) for 1 h were stimulated with IL-6 at a concentration of 10 ng/mL for 10 min to examine the inhibitory effect of LMT-28. As a result, pretreatment with LMT-28 was shown to inhibit the IL-6-induced p-STAT3 expression in a dose-dependent manner (Figure 1B).
Characterization of physicochemical properties of LMT-28
The typical physicochemical properties of LMT-28 including LogP and solubility were measured by using GLpKa (Pion, USA) based on D-PAS method and by 24 h shake-flask method based on equilibrium solubility, respectively [21]. The LogP value of LMT-28 was 3.65 and the solubility of LMT-28 in water was 0.39 mg/mL. Molecular weight of LMT-28 is 311.42, indicating that LMT-28 is very small molecule, compared to normal small molecular new drug candidates. LMT-28 is characterized by possessing one of hydrogen bond donor and five of hydrogen bond acceptors. Estimated PSA is 66.84 and number of rotatable bonds is eight. Predicted LogS is −2.96. These estimated structural physicochemical properties were predicted by Percepta software (ACD/Labs, Canada) and good values for new drug candidate by rule of five [22,23].
Metabolic stability of LMT-28
Metabolic stability in rat and human liver microsomes were measured up to 60 min as shown in Figure 2A, and metabolic stability values such as liver microsomal half-life, in vitro intrinsic clearance, and in vivo intrinsic clearance were summarized in Table 1. Liver microsomal half-life (t1/2) values of LMT-28 using rat and human liver microsomes were 15.3± 1.4 min and 21.9±2.8 min at the concentration 1 μM for rat and human, respectively. The calculated CLint,in vitro values were 91±8.4 and 64±8.1 (μL/min/mg protein) for rat and human, respectively. The CLint,in vivo values were also calculated by the equation in method section and those were 164±15 and 60± 7.7 (mL/min/kg body weight) for rat and human, respectively. These results suggest that LMT-28 undergoes metabolism in rat and human pooled liver microsomes at 10 μM and metabolic stability of LMT-28 in human may be higher than rat because of species difference.
In vitro pharmacokinetic study of LMT-28
Important in vitro pharmacokinetic factors which affect drug absorption and distribution such as permeability and plasma protein binding of LMT-28 are shown in Table 1. Accumulative amount of LMT-28 across MDCK cell monolayers were plotted at 30, 60, 90, 120 min after incubation in 12-well Transwell plate for transport study and shown in Figure 2B. The Papp of LMT-28 at 10 μM was 9.7±1.8×10−6 cm/s by cell permeability using MDCK cell monolayers. The plasma protein binding rate of LMT-28 at 2 μM was 92.4%±2.1% and mostly bound to serum albumin. These results suggest that LMT-28 may be absorbed and distributed easily with affordable permeability coefficient across cell membrane and acceptable plasma protein binding ratio in systemic blood circulation.
In vivo pharmacokinetics of LMT-28 in mice
After intravenous and oral administration at a dose of 5 mg/kg to male BALB/c mice (22 to 25 g), the mean plasma concentration-time curves of the LMT-28 are shown in Figure 3. Summarized pharmacokinetic parameters such as Tmax, Cmax, CL, Vss, AUC, MRT, and F are listed in Table 2. The pharmacokinetic values of CL, Vss, and AUC after intravenous administration of LMT-28 (5 mg/kg) were 8.66±4.51 L/h/kg, 12.9±4.66 L/kg, and 677±264 h·ng/mL, respectively. The pharmacokinetic values of AUC and Cmax after oral administration of LMT-28 (5 mg/kg) were 302±209 h·ng/mL and 137±100 ng/mL, respectively. These results indicate LMT-28 was orally exposed in mice.
Liquid chromatography-tandem mass spectrometry analysis of LMT-28 in biological fluids
The concentrations of LMT-28 in plasma or microsomal reaction solution were determined by LC-MS/MS. The LC-MS/MS analysis were performed by MRM mode using precursor ion and product ion mass spectra based on the chemical structure of analyte. LMT-28 and IS were analyzed for the abundant precursor ions [M+H]+ at m/z 312 and 237 and fragmented into produce product ions at m/z m/z 312 → 165 for LMT-28 and m/z 237 → 194 for carbamazepine, respectively. The chromatogram of LMT-28 and IS were eluted at 1.2 and 1.4 min with apparently sharp and symmetric peaks. A calibration curve for LMT-28 was generated by a linear least squares regression analysis by peak area ratio of LMT-28/IS chromatogram and the correlation coefficient (R2) ranged from 5 to 500 ng/mL to was greater than 0.99 with good linearity. LMT-28 was stable under various conditions of sample handling and processing.
DISCUSSION
LMT-28, a new drug candidate, with the mechanism of action as a novel therapeutic IL-6 inhibitor is under development. IL-6 inhibition by LMT-28 has been demonstrated to be effective in vitro and in vivo by previous reports [15] and confirmed once again by Western blotting (Figure 1). To develop LMT-28 as a candidate for a new drug, pharmacokinetic profiling is required for preclinical and clinical studies. Thus, in vitro and in vivo pharmacokinetic properties of LMT-28 were investigated and the pharmacokinetic properties of LMT-28 were confirmed in this study.
The physicochemical properties of LMT-28 were drug-like. The equilibrium solubility of LMT-28 was 0.39±0.12 mg/mL. This was slightly soluble in water system, however, it was a reasonable solubility for a new drug candidate. More importantly, new drug candidates may require a balance between hydrophilicity and lipophilicity [24]. Log P value of LMT-28 was 3.65±0.22 by GLpKa analytical system (Pion, USA). This Log P value indicated the lipophilicity of LMT-28 may be involved in high permeability across monolayer of MDCK cells.
In liver microsomal stability, LMT-28 was eliminated at a moderate rate by both human and rat cytochrome P450 metabolic enzymes. The half-life value of rat liver microsomal stability was 15.3 min and that of human was 21.9 min. In addition, there were interspecific differences between human and rat liver microsomal stability and metabolic stability of human liver microsome was slightly higher than that of rat. These data suggested the bioavailability in humans can be increased compared to that in rats. In addition, the differences between human and rat might be caused by the genomic differences of cytochrome P450 genes between human and rodent [25,26].
In this study, MDCK cells were used to examine cell per meability, drug absorption, and tissue disposition. Even though Caco-2 cell is one of the gold standard methods to evaluate drug properties, MDCK cells are also widely used for estimating both drug absorption and disposition to tissues [3,23]. In addition, compared to Caco-2 cells, MDCK cells have some advantages such as fast growing and no leakage of cell monolayer with tight junction expression and high TEER values (>700 ohms), which makes MDCK a good choice for estimating drug absorption and tissue disposition [27,28]. However, further evaluation of drug absorption may require Caco-2 cell transport studies.
LMT-28 had very good permeability properties in the MDCK cell permeability assay. The Papp was 9.7±1.8 (×10−6 cm/s) at 10 μM. Furthermore, in the plasma protein binding assay, LMT-28 showed moderate plasma protein binding properties (92.4%). These results suggested that most of LMT-28 bound to plasma protein for transport in blood circulation. Taken together, it is reasonable to suggest LMT-28 is a good drug candidate for novel therapeutic IL-6 inhibition.
In vivo pharmacokinetic studies using BALB/c mice, the area under the plasma concentration-time curve values of LMT-28 during 8 h (AUC8h) after oral administration (5 mg/kg) was 292±202 h·ng/mL and AUC∞ after integration into infinity was 302±209 h·ng/mL. The sampling time (8 h) was over 7 times of terminal half-life (t1/2; 1.13 h) and these AUC values for 8 hours (AUC8h) were able to cover approximately 96.7% of the total AUC (AUC∞). Thus, in the case of oral administration, the sampling time and the AUC value for 8 hours were sufficient to assess in vivo pharmacokinetics of LMT-28. The peak plasma concentration (Cmax) was 137±100 ng/mL and the time to reach Cmax (Tmax) was 0.80±0.67 h. After oral administration of LMT-28 to BALB/c mice, Cmax and Tmax showed good drug-like properties. Compared with the AUC value after intravenous administration, the bioavailability (F) of LMT-28 was calculated to be approximately 38.2%, and this may be regarded as a reasonable value considering that LMT-28 has been developed as a novel oral drug candidate for the first synthetic IL-6 inhibitor.
CONCLUSION
In summary, LMT-28 showed a moderate liver microsomal stability in both rat and human, and the bioavailability in human may be increased due to their interspecific differences between rat and human liver microsomal stabilities. In addition, the high cellular permeability of LMT-28 across monolayers of MDCK cells may be associated with sufficient exposure to systemic blood circulation following oral administration of LMT-28 in the in vivo system of mice. These results suggest that LMT-28 is a good oral drug candidate for the first synthetic IL-6 inhibitor.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print







