





INTRODUCTION
Circular RNAs (circRNAs) were first discovered in viruses [1], they attracted little attention until thousands of circRNAs were identified successively in mammals and humans [2,3]. CircRNAs were thought to be mis-splicing patterns, however, recently back-splicing was found to be ubiquitous in eukaryotes [4,5]. Some reports showed that circRNAs had biological function as miRNA sponges [6,7], regulated transcription [8] or bound to RNA binding proteins [5]. WhatŌĆÖs more, circRNAs were found to translate into polypeptides [9] and proteins [10], while extensive translation of circRNAs was driven by N6-methyladenosine [11].
Pigs are important meat producers. At the same time, pigs are one of the most appropriate animal models for human medical research, because of high similarity in physiological, anatomical, and biochemical, food structure and drug metabolism with humans [12]. Jinhua (JH) pig is a Chinese indigenous pig breed, being known for its slow growth and high fat deposition [13]. Conversely, the Landrace (LD) pig represents the fast growing lean type [14]. Distinct performance makes the two breeds ideal comparisons to study the impact of circRNAs on metabolism.
Liver is the most important organ of various metabolisms, playing a crucial role in the synthesis, decomposition, transformation, excretion and other metabolic processes. The study of specific circRNA function has been increasingly reported. The circRNA cZNF292 exhibits proangiogenic activities in endothelial cells in vitro [15]. CircHIPK3 regulates cell growth by sponging multiple miRNAs [16]. However, the research on circRNA in pigs is still relatively rare. Here we used male growing LD and JH pigs to describe the expression profiles of circRNAs with Ribo-Zero RNA-seq. In this study, we report the identification and characterization of circRNAs in the liver between the two distinct pig breeds. The intention of this study is to extend the circRNAs data in pig species and analyze putative circRNAs that may be related in breed-specific growth and fat deposition.
MATERIALS AND METHODS
Sample collection
Three uncastrated JH pigs were obtained from Jinhua Jianong Liangtouwu pig breeding farm (Jinhua, China) and three uncastrated LD pigs were purchased from Hangzhou Daguanshanpig breeding Co., Ltd (Hangzhou China). They were allowed free access to food and water ad libitum under controlled environment conditions. The experiment was approved by the Animal Care and Use Committee of Zhejiang University, Zhejiang, China (ZJU20160346). All procedures were carried out strictly with the guidelines and the regulations for the Administration of Affairs Concerning Experimental Animals of Zhejiang University Laboratory Animal Center. The right medial lobe of liver tissue were collected from the six pigs respectively at 70-day and immediately frozen in liquid nitrogen and stored at ŌłÆ80┬░C until RNA extraction.
RNA isolation
Total RNA was isolated from liver tissue samples using TRIzolTM reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturerŌĆÖs protocol. RNA quantity and purity were analyzed with the ND-1000 Nanodrop (Thermo Fisher, Wilmington, DE, USA) and RNA integrity number (RIN) was analyzed with the Agilent2200 (Agilent, Palo Alto, CA, USA) with RIN>7.0.
Library preparation and sequencing
Approximately 2 ╬╝g of total RNAs from each sample was used to prepare the library according to the following procedures: ribosomal RNAs (rRNAs) were removed from total RNA using Epicentre Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA, USA) and fragmented to approximately 200 bp. Subsequently, the purified RNAs were subjected to first strand and second strand cDNA synthesized following by adaptor ligation and enrichment with a low-cycle according to instructions of TruSeq RNA LT/HT Sample Prep Kit (Illumina, USA). The purified library products were evaluated using the Agilent 2200 Tape Station and Qubit2.0 (Life Technologies, Carlsbad, CA, USA) and then diluted to 10 pM for cluster generation in situ on the HiSeq2500 pair-end flow cell followed by sequencing (2├Ś100 bp) on HiSeq 2500 (Illumina, USA).
Data analysis
Quality of raw reads was evaluated by Phredscore33 (Q20, Q30). Adapter sequences and low quality reads were trimmed using Skewer and FastX-Toolkit respectively in raw data. The remaining clean reads were mapped to the reference porcine genome (S. scrofa 10.2) using TopHatv2.0.13. Then the unmapped reads were compared to fusion genes of reference genome using TopHat-fusion algorithm. Back-spliced junction reads which corresponded to splicing sites of exonic circRNA forms were selected. To annotate the origins of circRNAs, ANNOVAR was used to compare the location of circRNA splicing sites at both ends in the genome. The number of reads annotated in circRNA was counted in HTSeq v0.6.0. Based on the sequence predicting method, full-length circRNA sequence was predicted accordance to splice junction [6].
CircRNA conservation
Conservation analysis was used with phyloP (phylogenetic p-values) program base on conserved score of mammals obtained from the UCSC genome database. Each circRNA that met the conditions that conserved a score of more than 0.5 and was at least six bases length was then classified according to the position of acting element.
Differential expression analysis
The expression levels of circRNAs were calculated in reads per million reads (RPM). DEGseq, an R package, was applied to determine differentially expressed circRNAs. |log2 (Fold change)|>1 and q<0.05 were set as the threshold for significance. Hierarchical clustering was run to visualize differential expression patterns among samples.
Bioinformatic analysis and target prediction
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis for host genes of differentially expressed circRNAs were performed using KO-Based Annotation System (KOBAS) v2.0 [17], considering a corrected p-value <0.05 as significantly enriched. To analysis miRNA-circRNA interactions, the computational target prediction algorithms (miRanda 3.3a) was used to identify miRNA binding site.
Quantitative real-time polymerase chain reaction of different expressed circRNAs
Quantitative real-time polymerase chain reaction (PCR) was performed on an ABI Step One Plus system (Applied Biosystem, Carlsbad, CA, USA) using GoTaq qPCR Master Mix (Promega, Madison, WI, USA) with specific primers. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was chosen as a control to normalize the technical variations. The method of 2ŌłÆ╬ö╬öCt was used to calculate fold changes of circRNA expression. All reactions were run in triplicate. Statistical differences between the two breeds were performed by the student t-test and considered significant at the 0.05 level.
RESULTS
Characterization of liver tissue transcripts
Total RNA was depleted of rRNAs and linear RNAs prior to sequencing, however, there was still a few rRNAs fragments left in the process of creating a library. In order to avoid the influence of rRNA sequences, rRNAs were removed by comparison to the known sequences of rRNAs in the RNA central Consortium [18]. Raw data were processed to remove adapter sequences and low quality reads leaving 486 million clean data. After 0.13% to 0.25% known rRNAs reads were depleted, we obtained 485 million effective reads using TopHat v2.0.13 and samtools 1.2. Approximately 80% of these reads were mapped to un-fused genes of the porcine reference genome (S. scrofa 10.2) using Tophat2, while about 30% unmapped reads were aligned to fusion genes (Table 1).
Identification and annotation of circRNAs in liver tissue
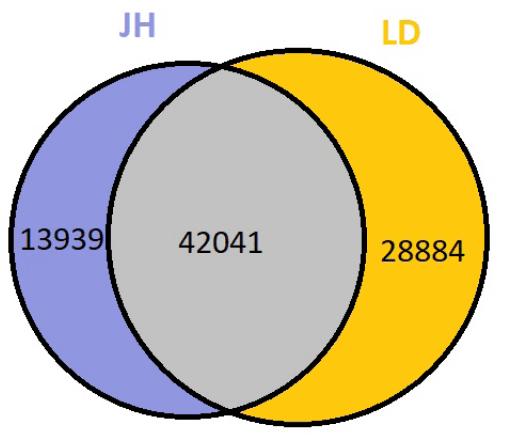
Specific alignments and corrections were applied to define putative circRNAs in the RNA-seq reads [19]. After screening and filtering, 0.48% to 0.81% of candidate back-spliced junction reads were assembled by TopHat. Based on the sequence characteristics of splice site, 0.28% to 0.52% of clean reads respectively in each individual were realigned to determine circRNAs (Table 2). We annotated these circRNA candidates based on the NCBI database and UCSC genome browser (Supplementary Table S1) and identified 84,864 circRNAs in two breeds (Figure 1). More than 86% of circRNAs consisted of exons while nearly 10% originated from introns and intergenic regions.
Differential expression analysis
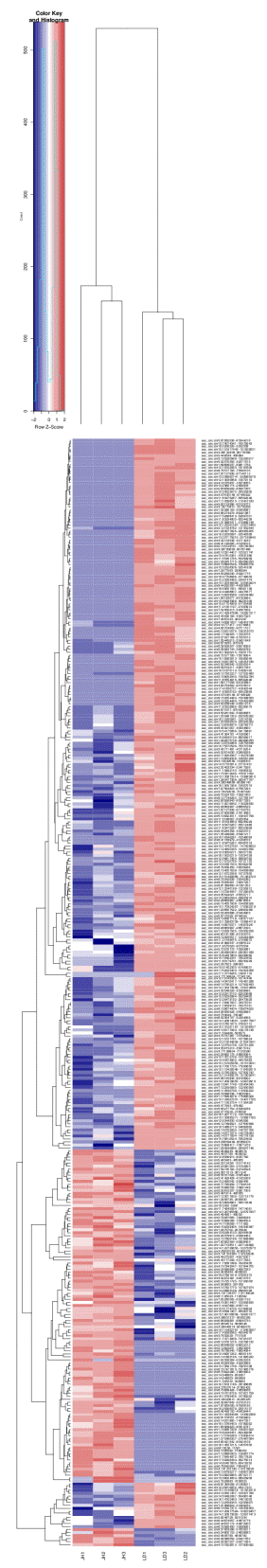
Differentially-expressed circRNAs in the liver tissues of LD (control group) and JH pig breeds were acquired by DEGseq. There were 116 up-regulated circRNAs in Landrace pigs compared with Jinhua pigs, whereas 250 circRNAs were downregulated (|log2(Fold change)|>1 and q<0.05). The heat map displayed circRNA (Figure 2) significantly differentially expressed in the two breeds (Supplementary Table S2). Among the up-regulated circRNAs, ssc_circ: chr4: 107829810-10783 0092 has the highest expression in JH pigs, while in the down-regulated circRNAs, ssc_circ: chr14:120977594-120979457 has the highest expression level.
Gene ontology enrichment analysis and Kyoto encyclopedia of genes and genomes pathway analysis of host genes
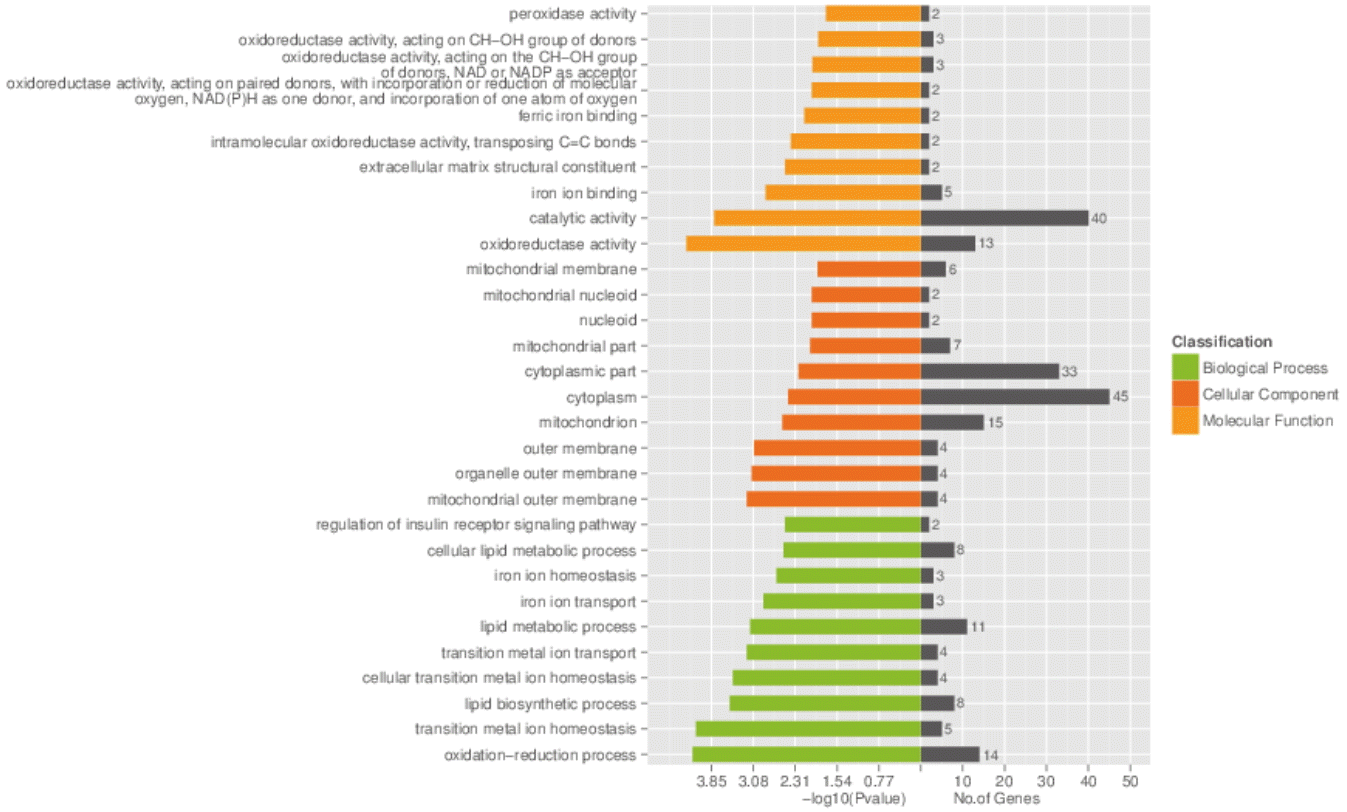
The GO analysis was applied to the host genes which differentially expressed circRNAs to explore the biological functions between two pig breeds (Figure 3). The figure shows GO enrichment for the host genes in biological processes, cellular components and molecular function sorted by p-value in an ascending order. The enriched GO targets of the host genes between JH and LD mainly differ in catalytic activity, cytoplasm and oxidation-reduction process.
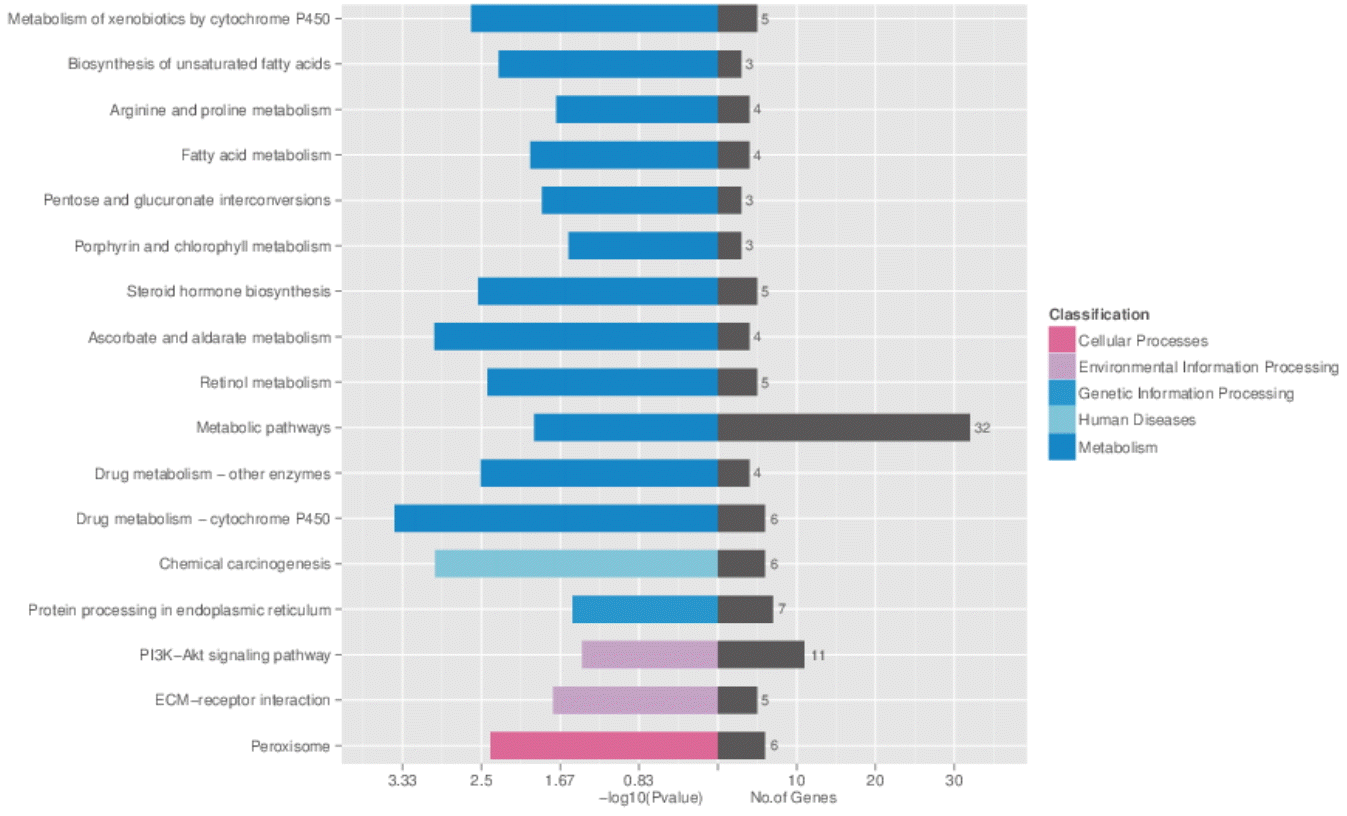
The host genes were classified according to KEGG function annotations to identify the pathways (Figure 4). The involved pathways included: metabolic pathways, PI3K-Akt signaling pathway, protein processing in endoplasmic reticulum, Peroxisome, drug metabolism (cytochrome P450) and biosynthesis of unsaturated fatty acids.
CircRNA conservation analysis
More than 58% differentially expressed exon-intron circRNAs and intergenic circRNAs have more than one block, however, the ratio in exon circRNAs to intron circRNAs is approximately 30%. Ssc_circ:chr6:35124281-35174142 has maximum blocks with six bases or more over 0.5 conservation score. The longest single block contains 95 consecutive bases belonging to exon-intron circRNAs and exon circRNAs had a higher proportion of conserved bases than circRNAs from other positions (Supplementary Table S3).
miRNA-circRNA interaction analysis
Free energy refers to the energy released when circRNA and miRNA combine and form a stable structure. The larger absolute value of free energy indicated the stronger binding ability. The total matching score of ssc-miR-30c-5p and circ:chr5: 105610075-105629122 was the highest, and also had maximum free energy at the same time. Ssc-miR-184 and ssc_circ:chr7:53408460-53472958, ssc-miR-140-5p and ssc_circ:chr2: 162425884-162509599, ssc-miR-122 and ssc_circ:chr2:162425884-162509599, ssc-miR-30c-5p and ssc_circ:chr5:105610075-105629122 also had high scores and free energy according to the prediction results of miRanda (Supplementary Table S4).
Quantitative real-time PCR validation
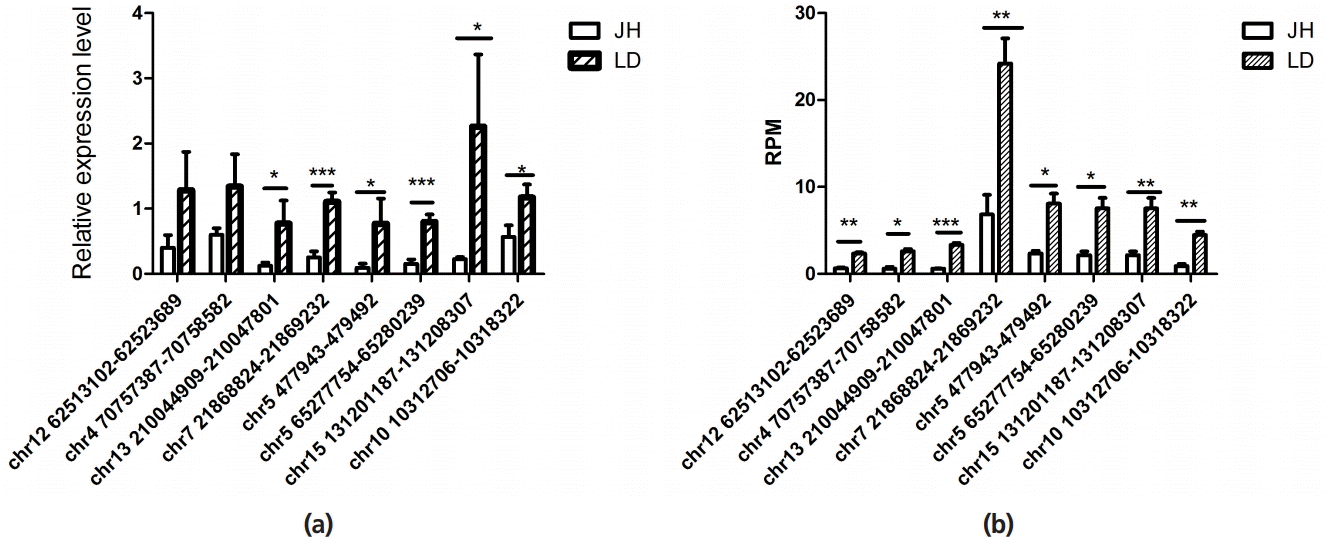
To validate reliability of RNA-seq results (Figure 5A), eight differentially expressed circRNAs were randomly selected and subjected to quantitative real-time PCR (qRT-PCR), with three biological replicates for each circRNAs of each breed (Figure 5B). The expression results indicated that the differentially expressed circRNAs with RNA-seq were reliable.
DISCUSSION
The liver plays a major role in carbohydrate, protein, amino acid and lipid metabolism [20]. In this study, we conducted a primary investigation of circRNA expression profiles in the liver of LD and JH pigs to detect the potential interaction with miRNA related to porcine growth and fat deposition. Ribosome RNA and linear RNA sequences were first removed to detect porcine liver transcriptome. Besides, toexclude the influence of the remaining rRNAs, we removed the rRNAs sequences by referring to RNAcentral Consortium [18]. Most of the 486 million reads obtained were mapped to the porcine reference genome (S. scrofa 10.2) by two comparisons. However, a certain number of clean reads (11% to 17%) were not mapped to annotated loci, which needs to be improved.
An algorithm method to identify circular splicing sites is more a efficient and unbiased way to seek circRNAs [4,7]. We identified 84864 circRNA candidates in two breeds. This number is much higher than the circRNAs in the porcine embryonic brain [3]. The reason for this drastic difference may be the circRNAs temporal and tissue-specific expression [3]. Because of the incomplete genome information in the untranslated regions, genomic origins of circRNAs candidates were only divided into exonic, intronic, intergenic, and other regions. In our study we observed that intergenic and intronic circRNAs were less conserved, according to the conservation score. Memczak et al [6] reported that circRNAs coding sequence was highly conserved, especially in the third codon position.
We obtained 366 circRNAs transcripts which were significantly differentially expressed in liver between the two breeds, and they might have a certain effect on RNA regulation layer [7]. In accordance with this, KEGG pathway analysis demonstrated that host genes of these differentially expressed circRNAs were associated with metabolism that may be related to liver function, such as the metabolic pathway. The results also showed that fat-metabolism-related genes were significantly different between LD and JH pigs. For example, stearoyl-CoA desaturase is a key rate-limiting enzyme in the synthesis of unsaturated fatty acids by insertion of a cis-double bond in the Delta9 position of fatty acid substrates [21]. Peroxisomal trans-2-enoyl-CoA reductase was reported to be a key enzyme of fatty acid metabolism as it catalyzes the reduction of unsaturated enoyl-CoAs to saturated acyl-CoAs [22]. Acyl-CoA synthetase long-chain family member 1 has demonstrated roles in animal fat synthesis and fatty ╬▓-oxidation and might be associated with fat deposition and meat quality traits [23]. Cytochrome P450 family 51, farnesyl diphosphate synthase, aisopentenyl-diphosphate delta isomerase 1 are also reported to play a role in cholesterol synthesis [24ŌĆō26]. In our study, eleven host genes of differentially expressed circRNAs were involved in lipid metabolic process and eight were enriched in lipid biosynthetic processes. Previous studies have determined that there were significant differences in fat deposition of the two breeds [13].
MiRNA plays a key role in development and specific biological processes, such as cell proliferation, differentiation, and apoptosis [27]. Previous studies showed miRNAs play important regulatory roles in the liver [28] and circRNAs acted as efficient miRNA sponges [7]. Memczak et al [6] has found that a human circRNA, antisense to the cerebellar degeneration-related protein 1 transcript, harbors 63 conserved binding sites for the ancient miRNA miR-7. CircHIPK3 was observed to sponge 9 miRNAs with 18 potential binding sites and specifically directly bound to miR-124 and inhibited miR-124 activity [16]. The circRNA HIPK2 regulates astrocyte activation via cooperation of autophagy and endoplasmic reticulum (ER) stress by targeting MIR124ŌĆō2HG [29]. According to analysis results of miRanda, there are multiple binding sites of ssc-miR-122 on ssc_circ:chr2:162425884-162509599. Mir-122 is highly abundant and acts as a regulator of cholesterol and fatty-acid metabolism in the liver [30]. These prediction binding sites implicate circRNAs as a miRNA sponge in the liver and suggest that circRNA may be an attractive regulatory in metabolic process.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement
Supplement Print
Print







