




INTRODUCTION
Meat quality is one of the most important aspects of consumer preferences, and the swine industry has recently focused on improving quality to ensure the profitability of the industry. The quality of meat is commonly determined by measuring pH, water-binding capacity (drip loss), color, and amino and fatty acids compositions within the loin (longissimus dorsi) muscle (LDM) [1]. These traits are related to tissue metabolism from the expression of multiple genes. Thus, there has been a strong drive for understanding the underlying regulatory mechanisms of gene expression in LDM.
DNA methylation, an important epigenetic modification of many eukaryotic genomes, is thought to play a major role in regulating transcription and/or translation of genes and the manifestation of metabolic traits [2]. DNA methylation controls a wide range of cellular functions and pathologies, including tissue-specific expression of genes, differentiation of cells, genomic imprinting, X chromosome inactivation, regulation of chromatin structure, carcinogenesis, and aging in mammals [3]. Different patterns, or levels, of DNA methylation have been observed in various tissues and also under different functional statuses of the same tissue [3]. The underlying mechanism of how exactly DNA methylation regulates expression of a gene still remains to be elucidated; the analysis of genome-wide DNA methylation patterns in various tissues is a helpful and powerful approach for understanding the relationship between tissues-specific regulations of gene expressions [4].
Genome sequencing following bisulfite conversion, methylated DNA immunoprecipitation (MeDIP), or methylation-specific polymerase chain reaction has recently enabled easier and more accurate identification of genome-wide DNA methylation patterns within a specific tissue in a high-throughput manner. MeDIP implements methylated cytosine-specific antibodies, thereby immuno-capturing methylated fractions of the genome using either array-based (MeDIP-Chip) or sequencing-based analysis (MeDIP-seq) [5]. Despite limitations in the MeDIP technology, including lower resolution and difficulty in discriminating between CpG and non-CpG methylation in single-end short reads, MeDIP-seq has been widely used to generate unbiased, cost-effective, genome-wide methylation profiles [6].
In livestock animals, only a few studies have investigated genome-wide DNA methylation profiles [7]. Li et al [8] reported that global DNA methylation of the pig genome associated with obesity systemically differed by breed, sex, and tissue. Yang et al [4] also found different methylation levels at CCGG sites in seven tissue genomes of pigs.
However, neither a comprehensive, genome-wide DNA methylation profile nor its functional inference in LDM tissue has been investigated. Therefore, this study aimed to identify and characterize the genome-wide DNA methylation profile in LDM of swine using MeDIP-seq.
MATERIALS AND METHODS
This study did not require animal use protocols or Animal Care and Use Committee approval as it only analyzed samples collected from commercial breeding farms after slaughter.
Samples and DNA preparation
The loin muscle samples from eight (four pairs of littermates) Duroc gilts, slaughtered at average body weight of 101.6┬▒5.3 kg and average age of 184┬▒9 days, were collected from four commercial breeding farms in Korea (NH, Kochang; Wonsan, Geochang; NIAS, Cheonan; Samsung, Eumsung). A pair that showed a largest difference in postmortem pH within a litter was selected from each farm. Each pair were fed on the same diet in similar environmental conditions.
The genomic DNA of samples was extracted and isolated using the phenolŌĆōchloroformŌĆōisoamyl alcohol method. Briefly, muscle tissue was frozen in liquid nitrogen, crushed with a mortar and pestle, and digested for 12 h at 55┬░C in a lysis buffer and 0.1 mg/mL of proteinase K. After incubation, 5 mL of protein precipitation buffer was added to precipitate proteins without heating. After centrifuging the solution at 13,000 rpm for 10 minutes, the supernatant was collected in a clean tube and 5 mL of phenol-chloroform-isoamyl alcohol (25:24:1) was added. After centrifuging at 13,000 rpm for 15 minutes, 5 mL of the aqueous phase was transferred to a clean tube, and the DNA precipitated with 0.5 mL of 3 M sodium acetate and 10 mL of 100% ethanol. Pellets were washed with 70% ethanol, allowed to dry, and dissolved in Tris-EDTA (TE) buffer.
Methylated DNA immunoprecipitation sequencing
For each sample, 1.2 ╬╝g of genomic DNA was randomly sheared to 200 to 300 bp fragments using a Covaris S2. MeDIP libraries were constructed using TruSeq DNA Sample Prep kit (Illumina, San Diego, CA, USA) following the manufacturer recommended protocols. DNA fragments were end-repaired, phosphorylated, polyA-tailed, and then ligated with Illumina single read adapters. Adapter-ligated DNA fragments 250 to 300 bp in size were screened using gel electrophoresis and purified with MinElute Gel Extraction kit (Qiagen, Valencia, CA, USA). Fragments were then used for MeDIP enrichment using Methylated DNA Immunoprecipitation kit (Diagenode, Li├©ge, Belgium) following the manufacturer recommendations. Immunoprecipitated DNA fragments were then PCR amplified: 98┬░C for 30 s; 10 cycles of 98┬░C for 10 s, 60┬░C for 30 s, and 72┬░C for 30 s; 72┬░C for 5 min. MeDIP libraries were quantified using Quant-iT Pico Green dsDNA Assay Kit (Ingen, Carlsbad, CA, USA). Flow cells were prepared with 8 pM DNA using the manufacturer recommended protocols and sequenced on an Illumina Genome Analyzer II to generate single-end 36 bp reads.
Identification of putative methylated regions
A generalized scheme of the experimental approach used to identify genome-wide putative DNA methylation peaks in the LDM of swine and the subsequent analyses are presented in additional file 1: Supplementary Figure S1 of the raw reads generated from MeDIP-seq, adapters, unknown, and low quality bases were filtered out by SolexaQA package (Phred quality score >25). For each LDM sample, the filtered MeDIP-seq reads were mapped to the NCBI pig reference genome build Sscrofa 10.2 (ftp://ftp.ncbi.nlm.nih.gov/genomes/Sus_scrofa/) by the Burrows-Wheeler alignment (BWA) (http://bio-bwa.sourceforge.net/), with less than 2 bp mismatches allowed.
Immunoprecipitated single-end sequence data were analyzed using MACS (release 1.4.2, http://liulab.dfci.harvard.edu/MACS/) to find genomic regions that were enriched for specifically precipitated DNA fragments. The browser extensible data (BED) files generated by MACS were then merged and analyzed by MACS to generate peak summit coordinates. Genome-wide putative methylated regions (PMR) in the LDM of swine were identified in a relatively conservative way based on biological replications. We defined PMR as methylated regions that overlapped more than 5% of at least two tissue samples. This minimized false-positives and increased the accuracy identified DNA methylation regions in LDM. Subsequent analyses were conducted using these PMR. In addition PMR that overlapped in all eight samples were defined as conserved methylated regions (CMR).
Data analysis
We performed clustering analysis using the hclust function in R (http://www.r-project.org/) to evaluate the effect of heredity on the variation in methylation patterns of individuals. The dissimilarity matrix was generated by calculating simple matching coefficients [9] based on the number of methylated regions in each individual sample that overlapped with PMR by more than 5%.
The relative distribution of methylation peaks among different genomic regions was determined from the genome annotations of the NCBI pig reference genome (Sscrofa 10.2) using BEDTools (version 2.1.7) [10]. Conservation was defined as the percentage of PMR observed in all eight samples within each genomic region. Genomic regions were classified nonexclusively into coding gene bodies (i.e., promoter, untranslated region [UTR], exon, and intron), types of repeats (e.g., long interspersed nuclear elements [LINE], short interspersed elements [SINE], long terminal repeats [LTR], simple repeats), cytosine-phosphate-guanine island (CGI), and intergenic regions. Annotation data for genomic elements were downloaded from the NCBI FTP site on July 21, 2013. Promoter regions were defined as 2 kb upstream of the transcription start site of a gene.
The functional enrichment of protein-coding genes containing PMR within the gene body (UTR, exon, and intron) was also analyzed to test whether there were biological functions (i.e., gene ontology [GO] terms and Kyoto encyclopedia of genes and genomes [KEGG] pathways) that were significantly related to the genes with PMR. For this, the list of 12,899 one-to-one human orthologs of pig protein-coding genes, among which 5,235 genes contained PMR, was obtained from the Ensembl database using BioMart. Functional enrichment of these genes was analyzed with all of the human orthologs as a background using the database for annotation, visualization and integrated discovery (DAVID). Significant enrichment of GO terms was assumed if the expression analysis systematic explorer (EASE) value was less than 0.05. To identify significant and relevant functional enrichment, the functional annotations of GO terms were filtered using a stringent criterion of EASE value <0.01 and a fold enrichment >1.3. For the KEGG pathway analysis, EASE value was <0.05 and fold enrichment was >1.3.
RESULTS
DNA methylation profile of the LDM
A total of 8.99 Gb raw reads were obtained from the LDM tissue of eight (four pairs of littermates) Duroc pigs using MeDIP-seq. After removing reads showing low-quality scores (Phred quality score <25), 8.26 Gb (92.1%) trimmed data were aligned to the NCBI pig reference genome build Sscrofa 10.2, and among these 7.06 Gb (85.4%) were annotated to the reference genome (Supplementary Table S1). As a result, in terms of the number of reads (read counts), an average of 25,685,050 trimmed read counts from each LDM sample were mapped to the reference genome.
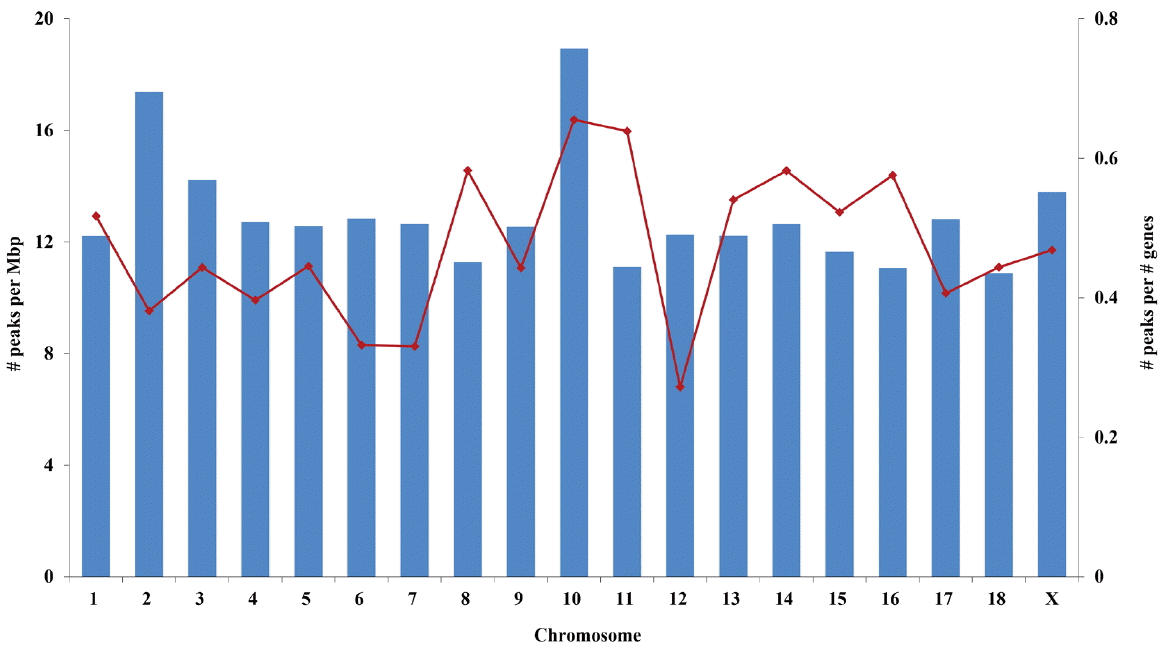
We detected methylation peaks along autosome and sex chromosomes in all eight samples using a MeDIP-seq analysis tool, MACS. Average length of the DNA methylation peaks in each sample ranged from 97 to 106 bp. The average number of genome-wide DNA methylation peaks in the porcine LDM sample was 93,939, and ranged from 68,185 to 133,663 (Supplementary Table S2). The highest average number of methylation peaks was in chromosome 1, whereas the lowest was in chromosome 12. A range of 33 to 40 methylation peaks per 1 Mb was identified for each chromosome. The highest density was observed in chromosome 10, while the lowest was in chromosome 12. The coefficient of variation of the number of methylation peaks among the samples for each chromosome was highest in chromosome 12 (6.6%) and lowest in chromosome 4 (0.8%).
To identify and characterize the signature of DNA methylation in porcine LDM, we used a relatively conservative method to define methylated regions among biological replicates. A total of 33,506 PMR were defined, and only 3.1% of these (1,029) were CMR (Supplementary Table S2). This implied that the level of DNA methylation conservation in the LDM of pig was low.
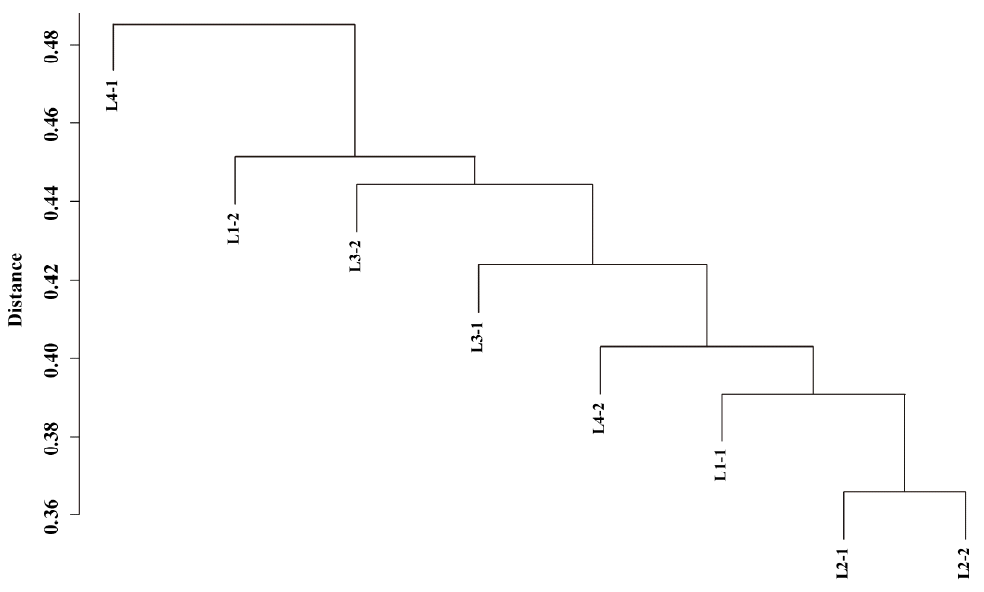
We also tested if the DNA methylation patterns were less variable between littermates. Hierarchical clustering of methylation peaks showed diverse genome-wide methylation patterns in each individual sample (Figure 1), and no structured similarity in methylation peaks between littermates was observed. Multivariate analysis of variance also showed no significant effect of litter on variation in methylation peaks (p = 0.47), so different DNA methylation patterns resulted mainly from individual variation.
Chromosome 10 contained the highest density of PMR on the basis of both chromosomal length and number of genes (Figure 2). Chromosomes 18 and 12 showed the smallest number of PMR per chromosomal length and number of genes, respectively (Figure 2). Interestingly, chromosome 10 also had the largest proportion of PMR that were conserved in all eight samples (6.1% vs the average, 3.1%; Supplementary Table S2).
Distribution of DNA methylations among genomic regions
Distribution of DNA methylations among genomic regions was non-exclusively identified. Only the results from the analyses with the PMR are presented here, which are consistent with the analyses using the DNA methylation peaks in each sample (Supplementary Table S3, S4).
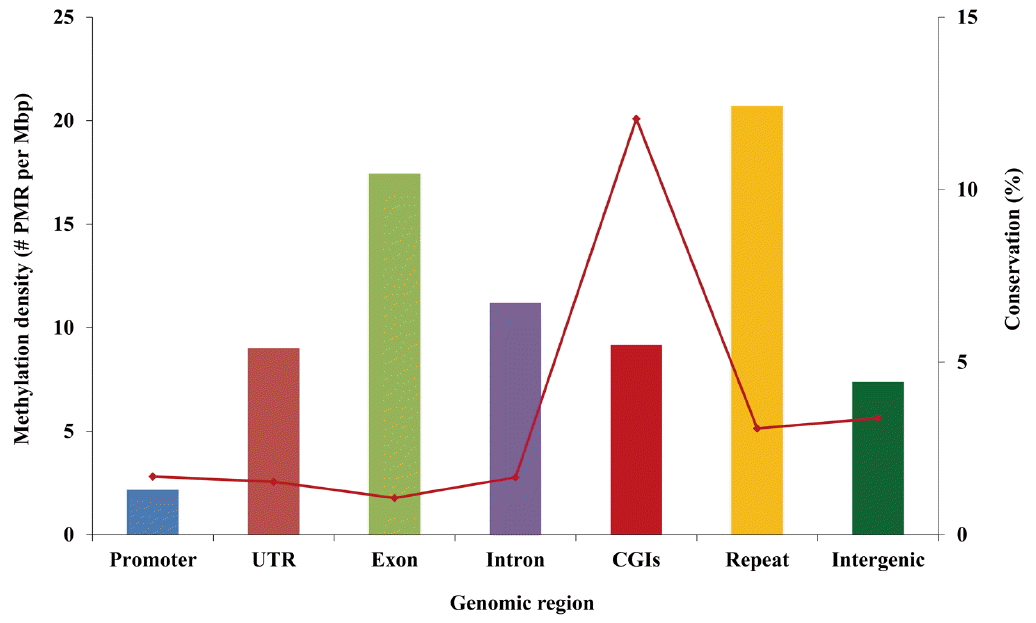
The relative density (the number of PMR per 1 Mbp) of PMR was highest in the repeats region (20.7 PMR/Mbp) across different genomic regions, followed by the exon region (17.4 PMR/Mbp) (Figure 3). The lowest density was observed in promoter regions (2.2 PMR/Mbp), followed by UTR (9.0 PMR/Mbp). Conservation of PMR was highest in CpG islands (CGIs, 12.1%) and lowest in exons (1.1%).
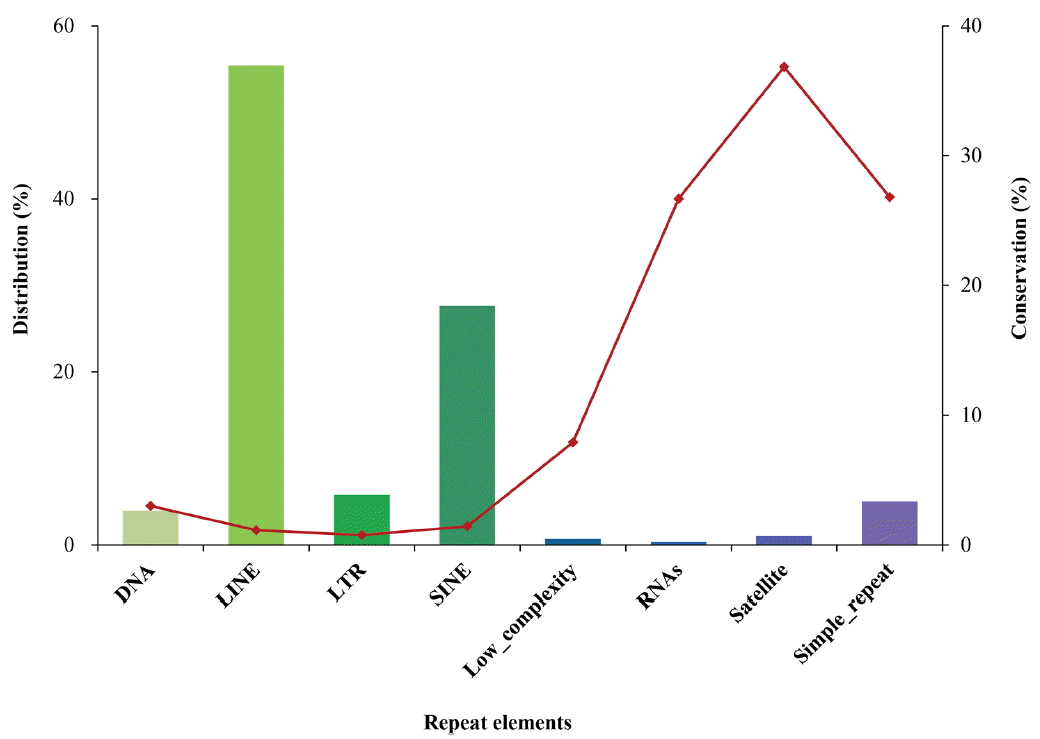
The PMR within repeats were further classified based on type (Figure 4). The majority of PMR within repeats were located in long (55.4%) or short (27.6%) interspersed repetitive elements (LINEs or SINEs, respectively), however, conservation of PMR among the samples was as low as 3.1%. Conversely, conservation of PMR located in satellite and simple repeats was as high as 36.8% and 26.8%, respectively, although relatively small numbers of PMR were found in these regions.
Distribution of DNA methylation peaks in CGIs
Methylations of highly conserved PMR in CGIs were further analyzed. There was a total of 477,585 CGIs based on the NCBI pig reference genome annotations (Sscrofa 10.2). An average of 1.2% was methylated in each sample, and most methylated CGIs were located in intergenic regions or introns (Supplementary Table S5). PMR were observed in 0.4% of CGIs, and the majority of CGIs containing PMR were located in intergenic regions (67.7%), followed by introns (24.0%) (Figure 5). Methylated regions were the most conserved in intergenic regions (20.2%). The lowest number of PMR and conservation was found in exonic CGIs. Although a relatively small number of PMR was found in the promoter CGIs, the level of conservation was relatively high (>16%).
Functional inferences of DNA methylation
Functional enrichment of the protein-coding genes containing PMR within their gene body (UTR, exon, and intron) was analyzed to assess functional inferences of DNA methylation. Among the 5,235 human orthologous genes contained PMR, a total of 3,595 genes were functionally annotated, and used for the functional analysis. Genes containing PMR were significantly (p<0.05) enriched for 14 KEGG pathways, 30 biological processes, 31 cellular components, and 15 molecular functions based on gene ontology (Supplementary Table S6).
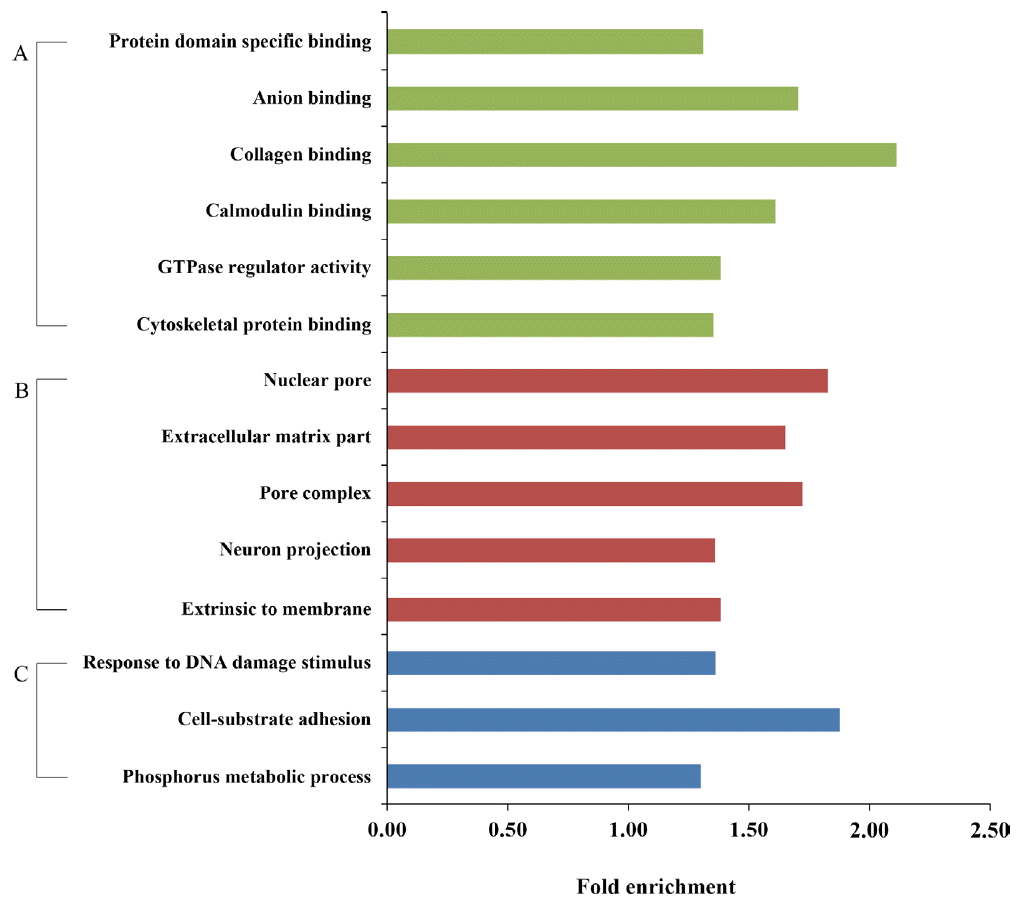
With more stringent criteria (cut-off p-value <0.01 and fold enrichment >1.3), we found that genes containing PMR were significantly enriched with the molecular functions of protein binding (e.g., collagen, anion, and calmodulin binding) and the biological processes related to cell-substrate adhesion, response to DNA damage stimulus, and phosphorous metabolic process (Figure 6). In addition, genes containing PMR were significantly more prominent in extra-membrane or pore complexes.
Thirteen out of 14 KEGG pathways that were significantly enriched for the genes containing PMR showed higher than 1.3 fold enrichment (Table 1). These pathways were related to muscular cell development, communication, cellular integrity and transport, and nutrient metabolism (e.g., insulin signaling pathway).
DISCUSSION
For decades, it was known that DNA methylation plays a central role in regulating gene expression and is essential for maintaining the normal biological functions of mammals [11]. To date, most studies have focused on development and cancer in mammals, while the functional role of DNA methylation in tissue specific regulation of gene expression, in relation to expression of metabolic traits, remains to be elucidated. In this study, we profiled genome-wide DNA methylation in the porcine LDM of four pairs of littermates of Duroc gilts, a breed known for high meat quality and lean growth, by analyzing next generation sequence MeDIP data. Unlike other recent genome-wide methylation studies in pig [8,12], which examined the level of DNA methylation from the number of mapped reads, we focused on the methylated regions along the chromosomes, and their diversity, conservation, and functional inferences among individual samples. These methylated regions may be related to the genetic machinery of gene expression and metabolic regulation, and therefore are possibly targets of interest for further studies on developing markers and deciphering the regulatory mechanisms of expression of metabolic traits.
DNA methylation in LDM may vary more by environmental factors than genetic factors. We found that DNA methylation patterns between two littermates were as diverse as between unrelated individuals. Hierarchical clustering from matching coefficients, and statistically supported by non-parametric multivariate analysis of variance, showed that DNA methylation of LDM was not clustered between two littermates. This implied that maternal genetic effects had little influence on the variation in DNA methylation patterns of individuals. Low heritability of DNA methylation may be due to variation caused by other environmental factors (e.g., in utero, age, diet, and exercise). This concurs with several studies that estimated the heritability of DNA methylation by comparing the epigenetic status of monozygotic and dizygotic twins. DNA methylation profiles were only minimally influenced by genetic variation [13,14]. An important factor that triggers changes in DNA methylation patterns, ŌĆśepigenetic driftŌĆÖ, is age [15] and age-related epigenetic drifts have also been reported in pigs [16]. Diet can also change DNA methylation pattern [17]. Although the littermates were fed on the same diet in similar environmental conditions, their DNA methylation was much varied. More research is needed to identify factors that affect DNA methylation patterns.
This study characterized a representative genome-wide DNA methylation map of the LDM of Duroc pigs, and analyzed the distribution of PMR among genomic regions. The highest density of PMR was observed in the repeat elements, as expected, and the second highest was observed in exons. Exons are hyper-methylated compared to introns in various species, which may be due to enrichment of nucleosomes that recruit DNA methyltransferases in exons [18]. The lowest density was found in the promoter regions. Hypo-methylation in the promoter regions is in accordance with other studies [7,12]; this phenomenon is well documented as it has a repressive effect on gene expression. Unlike DNA methylation in promoters, gene body methylation enhances the expression of genes [19]. This facilitating role of DNA methylation in the gene body can explain the relatively high level of DNA methylation within gene bodies and the positive correlation with gene expression.
Biased distribution of PMR in different repeat elements may be a species-specific characteristic. Our results showed that PMR among repeat elements were largely distributed in LINE and SINE, whereas only a few PMR were observed in RNAs. This is consistent with reported methylation patterns in pig liver [12]. Other studies in rat and chicken, however, reported the largest distribution of DNA methylation in Simple repeats and LTR among repeat elements, respectively [7]. Thus, species-specificity may be more important than tissue-specificity in the relative distribution of DNA methylation among repeat elements.
Our study indicated that most of CGIs were not methylated, but PMR in CGI showed a high level of conservation, which may be related to species- or tissue-specific gene expression. Methylation of intragenic or intergenic CGIs is known to play a critical role in regulating gene expression [20ŌĆō22]. CGIs in the promoter region are known to directly relate to the initiation of gene transcription [23], and methylation in CGI was negatively correlated with the level of gene expression [24] and related to repression of spurious transcription [19]. The low level of conservation, and low numbers of DNA methylation, in the exonic CGI suggested that methylation in the exonic CGI may not be a regular process. A relatively higher level of conserved methylated CGI in the genomic regions that regulate gene expression may indicate those methylated CGI are crucial for repressing spurious transcription and are related to cell protection and species- or tissue-specific characteristics.
Chromosome 10 had the highest relative density of PMR when the number of methylated regions was expressed either as size or as number of genes on each chromosome. Surprisingly, the gene density of chromosome 10 was lower than that of other chromosomes. Previous studies reported that the level of DNA methylation was highly correlated with gene density and the GC content of chromosomes [5,8]. The discrepancy between former studies and this one is likely due to the difference in methods of expressing DNA methylation levels, which in previous studies were defined by the number of mapped reads. This study used the number of methylated regions to focus more on the distribution of DNA methylated regions throughout the chromosomes. We conducted several different analyses (i.e., tests for biased enrichment of QTL and functional annotations using DAVID) to infer a possible reason for, or biological insights from, the high density of DNA methylation observed in chromosome 10. However, we could not find any significant functional signature in chromosome 10, and thus it is unclear what biological consequences may arise from the high density of DNA methylation in this chromosome. We speculate that DNA methylation in chromosome 10 may be related to essential biological functions or the normal functional integrity of LDM. The primary role of DNA methylation is to maintain normal biological functions by repressing spurious transcription of genes that are broadly expressed across tissues [19]. Further study, however, is needed to find conclusive evidence.
DNA methylation in the LDM was found to be related to specific biological functions and metabolic pathways. Functional enrichment analysis, conducted with a total of 3,595 orthologous genes containing PMR, indicated that these genes were significantly enriched with collagen, cytoskeletal protein binding, and cell-substrate adhesion, and were thus related to focal adhesion pathways. Focal adhesions are the specialized structures and areas formed by extracellular, transmembrane, and intracellular structures [25]. Adhesion proteins (e.g., fibronectin) are linked to other structures in an extracellular matrix, such as collagen and proteoglycans [25]. Focal adhesions play critical roles in the regulation of gene expression and the motility, proliferation, differentiation, and survival of cells [26]. In addition, genes containing PMR were also significantly enriched with functions related to nutrients metabolism, such as the insulin signaling pathway, starch and sucrose metabolism, and lysine degradation. The insulin signaling pathway is well known to control metabolism of various nutrients, such as carbohydrates and fats. Three genes (i.e., IRS1, IDE, and INSIG1) involved in the insulin signaling pathway contained PMR. Insulin plays a key role in muscle tissue by stimulating cell growth and differentiation, increasing glucose uptake, and enhancing protein and glycogen synthesis [27].
Some of the genes containing PMR were members of the insulin-like growth factor (IGF) family (i.e., IGF1, IGF1R, IGF2R, and IGFBP7) that influences secretion and action of insulin and insulin sensitivity of tissues [28]. Serum IGF-1 concentration is correlated with carcass traits. In pig, serum IGF-1 concentration was positively correlated with the intramuscular fat content at 8 weeks of age and the area of LDM at slaughter [29]. Another study observed that serum IGF-1 concentration and back fat thickness were positively correlated at the early stage of growth (6 weeks of age) and became negatively correlated at the late stage of growth (90 kg of body weight) [30]. Significant enrichment of insulin related metabolic pathways among the genes containing PMR suggested that DNA methylation may be partly responsible for determining the carcass quality of LDM.
CONCLUSION
In this study, we characterized a representative genome-wide DNA methylation map of the porcine LDM using MeDIP-seq. We found a relatively higher level of conserved methylation in genomic regions that primarily regulate the level of gene expression. This indicated that DNA methylation is crucial for repressing spurious transcription and is related to cell protection and species- or tissue-specific characteristics.
This study also showed that DNA methylation in the genome of porcine LDM plays an important role in regulating several biological process and metabolic pathways. Genes containing DNA methylation in the gene body were functionally enriched with cell development, cell-cell communication, cellular integrity and transport, and nutrient metabolism. Insignificant maternal genetic effects on variation in DNA methylation patterns suggested manipulating DNA methylation of porcine LDM may be possible via nutritional and environmental management.
The genome-wide DNA methylation map of porcine LDM from this study will provide a useful platform for further studies to decipher the epigenetic mechanisms of DNA methylation involved in expression of metabolic traits, and improve economically important carcass traits in pigs. Further research, however, is needed to investigate the direct and detailed mechanisms that underpin these processes.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement
Supplement Print
Print







