




INTRODUCTION
Myostatin (MSTN) is a member of super family of growth and differentiation factors (GDF-8), which is the only known inhibitor of skeletal muscle growth (Patel and Amthor, 2005). When MSTN activity is reduced or blocked, there is excessive growth of animal muscle with an increase in the number and diameter of muscle fibers. Natural MSTN mutations leading to “double-muscling” in mammals have been documented in sheep (Clop et al., 2006; Boman et al., 2009), dogs (Mosher et al., 2007) and cattle (Kambadur et al., 1997; McPherron and Lee, 1997). These mutations provided a potential to generate double-muscling trait in farm animals by knockout MSTN gene.
Although gene-targeted large domestic animals have been produced successfully with a combination of homologous recombination-based targeting and somatic cloning (McCreath et al., 2000; Lai et al., 2002; Richt et al., 2007), the rate of success is very low, and more importantly, disruption of the targeted gene is usually mono-allelic. It is prohibitively slow to obtain a null mutant. In addition, the lack of embryonic stem cells and difficulties of introducing large site-directed modifications hamper genetic engineering’s access to large animals. Zinc finger nucleases (ZFNs) are a powerful technology for genome engineering that allow researchers to induce subtle mutations. ZFNs are a class of engineered DNA-binding proteins that facilitate targeted genome editing by binding to a user-specified locus and causing a double-strand break (DSB), which can be spontaneously repaired by non-homologous-end-joining (NHEJ) in the absence of donor DNA. The NHEJ repair pathway may result in insertions or deletions at the targeted region, leading to a loss-of-function mutation (Zhang et al., 2014). The technology has been widely used for gene targeting in model organisms such as Drosophila (Bibikova et al., 2002), Arabidopsis thaliana (Lloyd et al., 2005), zebrafish (Doyon et al., 2008; Meng et al., 2008), mouse (Meyer et al., 2010), rat (Chu et al., 2012), silkworm (Bombyxmori) (Takasu et al., 2010), frog (Young et al., 2011), rabbit (Flisikowska et al., 2011), and large animals such as pigs (Hauschild et al., 2011) and cattle (Yu et al., 2011). Zhang et al. (Zhang et al., 2014) successfully produced sheep fetal fibroblasts with a MSTN disruption at the exon 1. However, there has never been a bi-allelic knockout of the MSTN gene in sheep.
In this study, we confirmed that ZFNs could recognize and cleave a target sequence in MSTN exon 3 in sheep primary cultured somatic cells and parthenogenetic embryos. The screening of single-cell colony by serial dilution avoided the use of antibiotic selection, and increased the biological safety. Our results suggested that ZFNs could be a useful tool in the production of MSTN knockout sheep.
MATERIALS AND METHODS
Design and construction of zinc-finger-nucleases
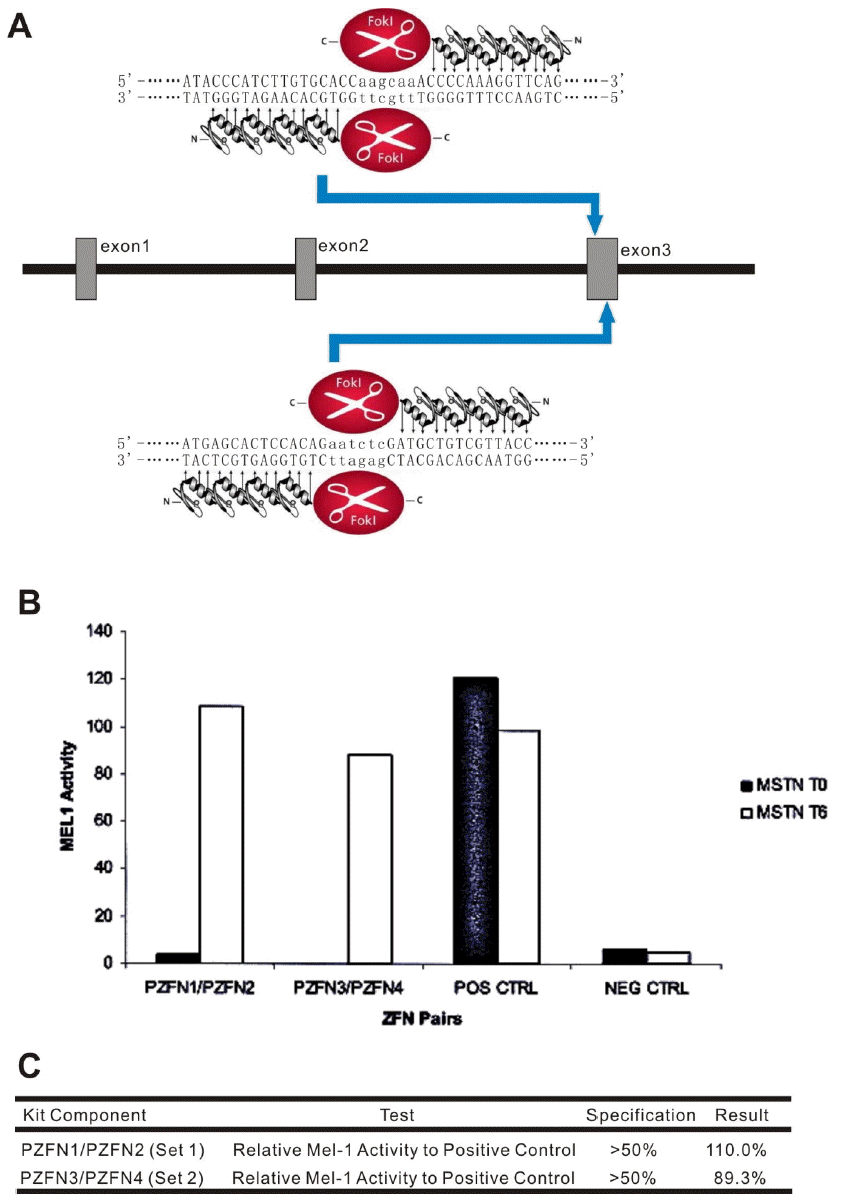
Two pairs of custom-designed ZFN plasmids (pZFN1/pZFN2[set1], pZFN3/pZFN4[set2]) and mRNA were designed and constructed by Sigma-Aldrich (St. Louis, MO, USA) to target the specific site of exon 3 of sheep MSTN. ZFN activity was measured with a yeast MEL-1 reporter assay (Doyon et al., 2008). Detailed sequence information of the zinc finger modules can be found in Figure 1B and 1C.
Cell culture and transfection
Sheep fibroblasts were isolated from male lambs three days after birth as previously described by Hu et al. (2012) with slight modifications. Ear tissues were sheared and soaked in D-Hanks supplemented with 100 μg/mL penicillin and 50 μg/mL streptomycin. Explants were poured into 35 mm culture dishes with D-Hanks containing penicillin and streptomycin. Connective tissue and fat were removed and the remaining tissue was soaked in 100 μL fetal bovine serum (FBS) and minced with sterile scissors. Tissue lumps were distributed evenly on 25 cm2 flask with tweezers and cultured at 37°C in an atmosphere of 5% CO2 for at least 12 h. Then 4 mL of fresh cell culture medium (Dulbecco’s modified Eagle media (DMEM), 20% [v/v] FBS, 100 μg/mL penicillin, 50 μg/mL streptomycin) was added and changed every three to four days. The cells were kept stationary for seven days. Flat and spindle fiber cells swam out after seven days, and when the cells from the explants reached 80% confluency, they were treated with 0.25% trypsin/0.05% EDTA (ethylene diamine tetraacetic acid) and passaged at 1:2 or 1:3. Then the cells were trypsinized and frozen in 20% FBS and 10% DMSO (dimethyl sulfoxide) for long-term storage.
Cellular transfection was performed using lipid-based transfection reagents. Briefly, the above isolated sheep fibroblasts were seeded at 5×105 per well in 6-well plates the day before transfection and allowed to replicate until they reached 80% to 90% confluency. The cells were transfected using lipofectamin 2000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. The medium was removed and the fibroblasts were washed twice by adding 2 mL of fresh, hot Opti-MEM (Life Technologies, USA) into the cells. The ZFN mixture was prepared by mixing the following reagents: 1 μg of pZFN1 plasmid, 1 μg of pZFN2 plasmid, 5 μL of lipofectamin 2000. After 20 to 25 min room temperature, the mixture was added into plates and mixed by shaking slightly. The plates were then placed in incubator with 5% CO2 at 37°C. After 3 hours transfection, the Opti-MEM was removed and the medium was replaced by DMEM with 10% FBS and the cells were cultured at 37°C for 48 hours.
Screening of single-cell colonies
When the cells reached 80% to 85% confluency forty-eight hours post-transfection, they were treated with 0.25% trypsin/0.05% EDTA and plated by limiting dilutions. Digested cells were counted and diluted at a density of 500 cells/10 cm2 in plate and the medium was changed every three days. After 7 to 9 days of drug-free culturing, the cell colonies were picked out with filter paper and placed in 24-well plates to expand the population. When cells were nearly confluent in the wells, they were harvested by trypsin digestion and divided into two parts. One part was expanded by culturing in 24-well plate for cryopreservation and the other part was used to identify targeting events.
Identification of mutated cells derived from single cell colony and parthenogenetic embryos
The genomic DNA was extracted from cells and blastocyst of parthenogenetic embryos for screening the targeting events using QIAamp DNA Micro Kit (QIAGEN, Frankfurt, Hessen, Germany). Briefly, the cells were washed 3 times in phosphate buffered saline (PBS) and digested with 0.25% trypsin/0.05% EDTA. After digestion, 500 μL PBS was added and centrifuged at 6,000 g (8,000 rpm) for 2 min. Cell pellet was resuspended and DNA was extracted according to the manufacturer’s instructions. DNA samples were used to amplify the target sequence with the primers of MSTN-F1(5′ GTGTCAGGCATTCAG ATATTC 3′), MSTN-R1(5′ GCTTGTGCTTAAGTGACT GTAGC 3′) according to the following program: 95°C for 5 min followed by 95°C for 30 s, 61°C for 30 s, 72°C 45 s for 35 cycles, and ended by 72°C for 7 min.
For parthenogenetic embryos, each embryo was washed 3 times in PBS and DNA was extracted using QIAamp DNA Micro Kit (QIAGEN, Germany). A two-step nested polymerase chain reaction (PCR) was used to amplify the targeted sequence. In the first round PCR, a touch-down PCR with the primers of MSTN-F1/R1 was performed by 95°C, 5 min followed by 95°C, 30 s, 65°C to 53°C, −2°C/cycle, 30 s and 72°C, 45 s for 14 cycles. In the second round PCR, the first round PCR products were diluted 25 times. One microlitre of each PCR reaction and primer pairs MSTN-F2(5′ AGCGATAAACAAGACAAAGC 3′), MSTN-R2(5′ ATGAGCACCCACAGCGATCTACT 3′) were subjected to a second round of amplification by 95°C 30 s, 61°C 30 s, 72°C 45 s for 35 cycles, and terminated by 72°C for 7 min.
To identify indels, PCR products of cells and blastocyst were purified using a Gel Extraction kit (TianGen, Beijing, China) and the purified PCR products were sequenced directly using primers. For the double peaks in chromatograph, further sequencing was carried out by TA cloning sequencing (pMD-19T vector, TakaRa, Japan). Mutations were identified by alignment of sequenced alleles to wild type allele.
Cell viability assay
A cell viability assay was performed using the Cell Counting Kit (CCK-8 kit, Dojindo Laboratory, Kumamoto, Japan) according to the manufacturer’s specifications. Briefly, primary fibroblast cells and MSTN targeted cells were seeded at 2,000 cells per well in 96-well plates and counted for 6 days continuously at 24, 48, 72, 96, 120 h incubation by addition of 10 μL CCK-8 reagent to each well. For each counting, after 3 h of incubation at 37°C, the absorbance of each well was measured at 450 nm wavelength by a microplate reader (Thermo, Varioskan Flash, Waltham, MA, USA) and a cell growth curve was generated.
Microinjection of zinc-finger-nucleases mRNA into Sheep embryo
Collection, maturation and culture of sheep oocytes in vitro were carried out as described by Park et al. (2012). Briefly, ovaries were collected from a local abattoir and transported to the laboratory in sterile physiological saline at 37°C. Immature oocytes were aspirated from medium size (3 to 6 mm) antral follicles using a 9-gauge hypodermic needle attached to a 10 mL disposable syringe.
After 22 to 24 h maturation, sheep oocytes were freed of cumulus by treating the cultured cumulus-oocyte complexes with hyaluronidase (0.1%), and the matured oocytes with an extruded polar body were activated by adding into chemical reagents as follows: the matured oocytes were transferred into 5 μmol/L ionomycin for 5 min, and then the oocytes were washed 2 times with culture solution and 3 times with 2 mmol/L 6-Dimethylaminopurine (6-DMAP). Subsequently, the treated oocytes were incubated for 2 to 3 hours at 38.6°C in an atmosphere of 5% CO2 in air. The activated oocytes (parthenogenetic embryos) were subjected to cytoplasmic microinjection of ZFN mRNA (set 1) at 12 h post activation.
ZFN mRNA (set 1) was diluted with nuclease-free water at concentration of 25 ng/μL, 50 ng/μL, and 100 ng/μL in preparation for micro-injection. Different concentrations of mRNAs encoding a particular ZFN set were micro-injected into the cytoplasm of one-cell stage partheno-genetically activated sheep embryos using a Nikon micro-injection system under standard conditions. The cleavage rate of parthenogenetic embryos was calculated at 48 h post activation and blastocysts were harvested at 144 h post activation for MSTN targeting analysis.
RESULTS
Assessment of zinc-finger-nucleases activity in vitro
Two different ZFN pairs targeting the sheep MSTN exon 3 (Figure 1A) were obtained from Sigma Aldrich. ZFN activity was measured by the yeast MEL-1 reporter assay and cleavage activity was evaluated before (0 h, blue bars) and after induction of ZFN expression (6 h, red bars). MEL-1 levels were positively correlated with ZFN ability to create double strand breaks at the desired target site. The cleavage activity of ZFNs 50% higher than that of positive control after induction was chosen for genome editing. Two pairs of zinc finger modules displayed MEL-1 activity at 110.0% and 89.3%, respectively (Figure 1B and 1C). A ZFN mRNA recognized the site of aagcaa in exon 3 (set 1) showed higher ZFN activity and was used to for MSTN targeting.
Isolation and identification of zinc-finger-nucleases targeted cell lines
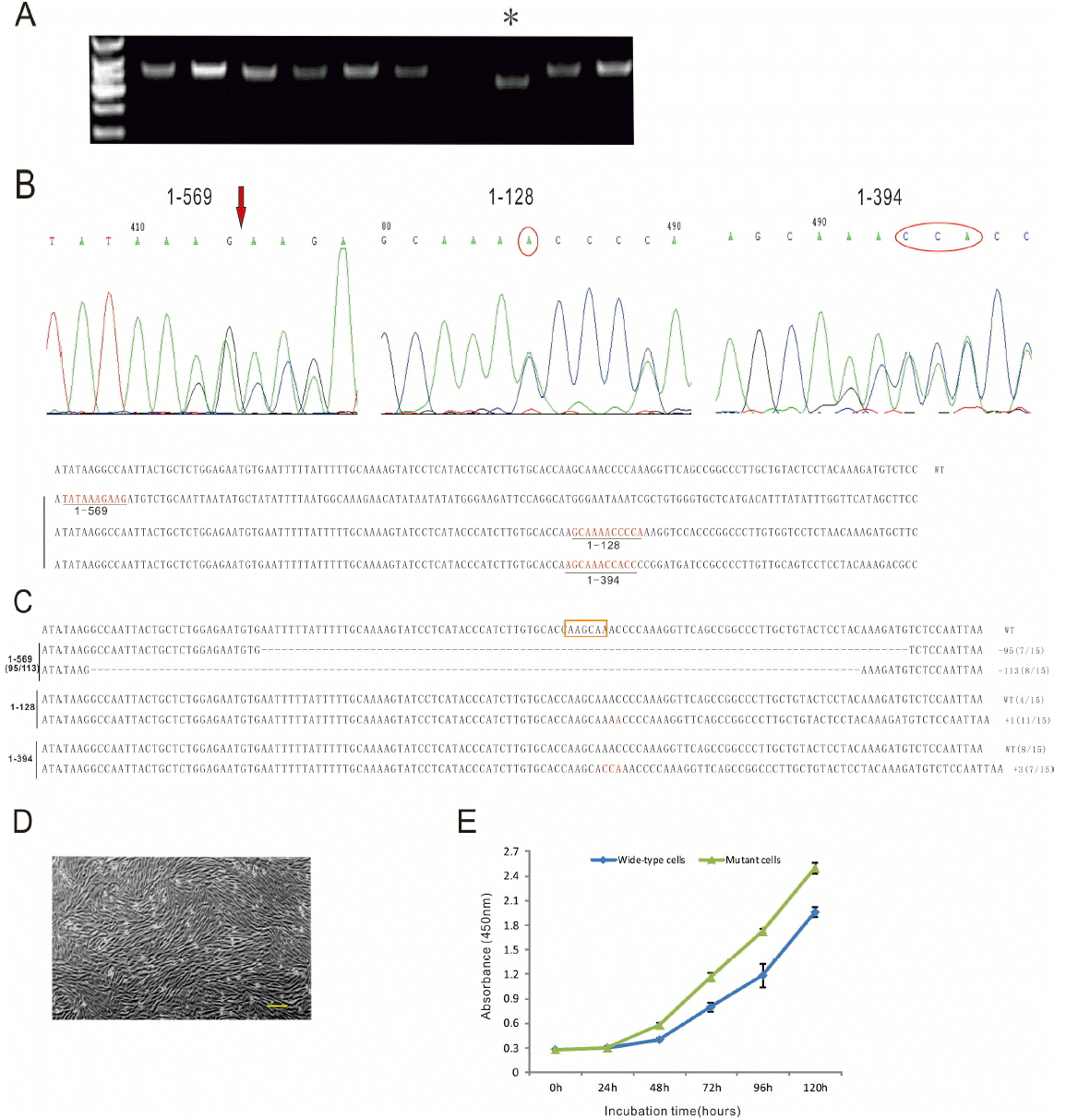
A total of 219 cell colonies were isolated and cultured for targeting screening by sequencing. An obvious short PCR product was observed in colony number: 1–569 (Figure 2A) and further confirmed by sequencing. Two targeted cell colonies with one insertion (A) at the position of 485 locus (colony number: 1–128) and three bases insertion (GCA) at the position of 493 locus (colony number: 1–394) were identified by sequencing (Figure 2B). Cloning sequencing to determine bi-allele or mono-allele targeting events showed that two colonies were identified with mono-allele mutation and one colony with bi-allelic deletion at the predicted locus (Figure 2C).
Interestingly, sequencing of 15 clones derived from 1–569 cell line displayed two deletions, with 113 bp deletion in one allele (8/15) and a 95 bp deletion in the other allele (7/15). No wild type allele was found in 1–569 cell clone. It indicated that 1–569 cell line was homozygote targeting cell. However, though two kinds of alleles were identified in 1–128 and 1–394 cell lines, only one mutation happened in an allele in each cell line. The other allele was wild type. In 1–128 cell line, 11 of 15 alleles were “A” insertion mutation and 4 of 15 alleles were wild type. Likely, in 1–394 cell line, 7 of 15 alleles were “GCA” insertion mutation and 8 of 15 alleles were wild type. The mutated alleles accounted for 73.3% and 46.7% in 1–128 and 1–394 cell line, respectively. It inferred that these cell lines were heterozygous.
Growth curve of targeting cells
Cell proliferation was monitored using the CCK-8 kit for primary fibroblast cells and MSTN targeted cells according to the manufacturer’s protocol. The optical density observed in MSTN targeted cells at 450 nm was significantly greater than that of primary fibroblast cells after two days culture (Figure 2E). MSTN targeted cells were characterized by a shorter doubling time and faster plateau phase compared to primary fibroblast cells (p<0.01, data not shown).
Zinc-finger-nuclease-induced MSTN mutations in sheep embryos
To test the effects of ZFNs on gametocytes, we examined ZFN-induced mutation in parthenogenetically activated embryos. Total of 144 oocytes were injected with ZFN mRNA. Among the injected embryos, 18 of 36, 27 of 85, and 7 of 31 embryos with injection of 25 ng, 50 ng, or 100 ng ZFN developed to blastocysts, respectively. Sanger sequencing of the 37 blastocysts identified 13 mutated embryos with two forms of 113 or 95 bp deletions, which was the same as that in cell lines (Figure 2C). The overall efficiency of deletion in embryos was 35% (13/37), and increased with the increase of ZFN mRNA dose. However, the blastocyst ratio was decreased with the increase of ZFN dose (Table 1), which inferred that the high dose of ZFN mRNA may affect the embryo development. Further T-A cloning sequencing evidenced the two deletions of 95 and 113 bp in embryos. Among the 35 sequenced colonies of 13 embryos, seventeen were genotyped as deletion (twelve clones with 95 bp deletion and five clones with 113 bp deletion) and eighteen were genotyped as wild type (Figure 3B).
DISCUSSION
Our study demonstrated the feasibility of a ZFN-based approach to rapidly target MSTN gene knockouts in sheep fibroblasts and embryos. In order to improve transfection efficiency, we tested different ratios of ZFN plasmid to lipofectin 2000 (μg/μL): 2:4, 2:5, 2:6, 3:6, and found there was no distinct difference in transfection efficiency (data not shown). Moreover, to evaluate ZFN targeting efficiency of cells, we extracted cell DNA and sequenced it. The results showed that there was no mutations in cells. The most probable reason was likely to be the low transfection efficiency which leads to few mutation cells being detected. The colonies derived from single-cells were identified by different indels, of which 1–128 clone had a 1-bp insert in exon 3 in MSTN gene leading to premature termination and 1–569 clone had a bi-allelic deletion of 113 bp in one chromosome and 95 bp in the other chromosome leading to a frame shift and truncated protein. We further carried out cell viability assays to investigate whether MSTN mutations could induce cell proliferation. Our results (Figure 2E) showed a substantial increase in the number of targeted cells compared with the primary fibroblast cells. The result indicated that 1–569 cell line could be used as donor cells for somatic cell nuclear transfer to generate MSTN targeted sheep.
ZFN-mediated genetic modification has been achieved in zebrafish, frogs, mice, rats and pigs by direct microinjection of ZFN plasmids or mRNA into a single cell embryo (Meng et al., 2008; Geurts et al., 2009; Cui et al., 2011; Young et al., 2011). However, studies on direct injection of ZFN mRNA into sheep embryos has not been reported in sheep. In this study, we microinjected different concentrations of ZFN mRNA into sheep parthenogenetically activated embryos. Interestingly, only 95 and 113 bp deletion forms were identified in blastocysts by PCR (Figure 3A) or clonal sequencing, which was consistent with the result of 1–569 cell clone. This result revealed that the ZFN-initiated double strand breaks and repair by the non-homologous end joining pathways might be identical for a certain ZFN module. Furthermore, the injection of high concentration ZFN mRNA into embryo resulted in high embryo mortality (data not shown), which indicated that high concentration ZFN could be harmful for embryo survival and development.
In the past three years, we performed targeted disruption of MSTN in sheep using ZFNs by two different approaches, nuclear transfer and direct embryo injection.
Colony 1–569 (95 and 113 bp deletions) was used as nuclear donor for somatic cell cloning. A total of 266 cloned embryos were introduced into 58 surrogate mothers. After full-term gestation, which lasted around 150 days, regrettably, none of the lambs were delivered.
Alternatively, ZFN-encoding mRNA (set 1) was microinjected into the cytoplasm of one-cell stage embryos which were obtained by Merino sheep superovulation according to the method described above. After the injected embryos developed to two-cell stage (~24 h, 37°C), 195 of the 226 injected embryos were transferred into 77 surrogate ewes. Of these, 13 pregnancies were established as determined by ultrasonic observation. After about 150 days, 15 lambs were delivered from these surrogate pregnancies. Sequence analysis revealed that none of them carried the predicted ZFN-induced mutation in their genome and all of them were parental genotypes.
Based on these results, we presume that although ZFN mRNA can effectively edit MSTN gene, the deleted locus may adversely affect further development of the embryo. In addition, although previous reports indicated that ZFN had high specificity, it also introduced multiple deletions in somatic cells and offspring derived from these cells. Moreover, only colonies with the desired bi-allelic short fragment deletion were used as nuclear donors.
In our results, the length of bi-allelic deletion fragments was as high as 100 bp. Although many studies have been carried out using ZFNs with a focus on cells and livestock using ZFNs, no study has been reported with offspring originating from colony cells with bi-allelic long fragment deletion (>100 bp). It is quite likely that bi-allelic long fragment deletion, epigenetics, chromosomal aberration in donor cells or off-target mutations by ZFNs affect genome replication and normal embryonic development.
ZFNs, transcription activator-like effector nucleases (TALENs), and clusteredregularly interspersed short palindromic repeats (CRISPR) technologies are transformative tools that have the potential to revolutionize biological research. Each of these technologies utilizes restriction enzymes to introduce a DNA double stranded break at a targeted location with the guide of homologous binding proteins or RNA. Nevertheless, ZFN technology is not convenient since ZFNs cannot be easily programmed for the recognition of new target sequences and it has high cost. Moreover, off-target effects may lead to the introduction of unintended mutations and cytotoxicity. In general, construction of ZFNs with high specificity is not simple compared with TALENs and CRISPR/Cas9. TALENs, whose construction is much easier than that of ZFNs, have higher genome editing activity and less toxicity than ZFNs. However, while the modular nature of TALE eases the difficulties of design and selection of specific DNA- binding modules, the large size of TALENs may limit their delivery by size-restricted vectors such as recombinant adeno-associated virus. The Cas9 endonuclease operates as a monomer to induce DSBs, whereas the FokI in ZFNs and TALENs operates as a dimer. The enzymatic machinery remains the same for any intended target; only the guide RNA provides DNA binding affinity and therefore targeting. Thus, CRISPR/Cas9 requires no protein engineering for any change in target, only synthesis of a new guide RNA. This simplicity has dramatically reduced the time needed to conduct genome engineering experiments. Compared to ZFNs and TALENs, CRISPR/Cas9-mediated genome engineering is easy to handle, highly specific, efficient, and multiplexable. Owing to its simplicity, convenience, and flexibility, CRISPR/Cas9 technology is currently evolving with astonishing rapidity. At present, because of the widespread application of the CRISPR/Cas9 technology, we didn’t continue to study the mechanism of the influence of MSTN on embryonic development by ZFNs. Earlier, we generated CRISPR/Cas9 mRNAs specific for sheep MSTN and microinjected them into the cytoplasm of ovine zygotes and successfully produced gene-modified sheep (manuscript currently under preparation).

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







