




INTRODUCTION
Domestic quails, derived from the Japanese quail (Coturnix japonica), as laying, meat, and laboratory animals have produced a flourishing industry in the world (Charati et al., 2014). At present there are about 1,050 million quails worldwide (Chang et al., 2007). Due to their known properties, quails have been widely used for biological and genetic studies (Alkan et al., 2013). Also, quail are considered an economically important avian species and provide an alternative to the more common chicken (Farrag et al., 2011).
Determination of the genetic diversity of domestic flocks is a necessity for providing needed information for the conservation of useful genotypes against future uncertainties such as global warming, diseases and humanitarian needs. Continued genetic improvement of livestock is dependent on the fact that substantial genetic variation exists within individual breeds allowing them to respond to selection for different traits (Farrag et al., 2011). Quantitative assessment of genetic diversity within and between populations is an important tool for decision making in genetic conservation plans (Davila et al., 2009).
Currently, many molecular marker methods have been developed to estimate genetic relatedness within and between livestock species. Microsatellites or simple sequence repeats (SSR) have emerged as one of the most popular genetic markers for a wide range of applications in population genetics, conservation biology, and evolutionary biology (Abdelkrim et al., 2009) and occur in both coding and non-coding regions of DNA. SSRs have high mutation rates and simple Mendelian inheritance which make them particularly suitable for the study of fine population structure, mating systems and pedigrees (Abdelkrim et al., 2009). Also, they are easy to genotype and are densely distributed throughout eukaryotic genomes, making them the preferred genetic marker for high resolution genetic mapping (Babar et al., 2012). Recent information in literature has revealed that microsatellite markers are not only useful in determining heterozygosity and estimating genetic distances among closely related species (Chen et al., 2004), but also are now one of the most widely used molecular markers, due to their relative ease of scoring and high levels of polymorphism (Gomes et al., 2013).
Anatolia is a natural habitat for quail and on the migration route of quail flocks. There are also commercial enterprises producing quail, especially Japanese quail. Quail eggs and meat are consumed extensively in Turkey and quails are also grown as ornamental animals. Although there are many studies on quail breeding and genetics, sufficient genetic information has not yet been obtained in Turkey. The objective of the present study was to investigate genetic differences among long term selected five Japanese quail lines using 15 microsatellite markers and discussion of selection effects.
MATERIALS AND METHODS
Five Japanese quail (C. coturnix japonica) lines were obtained from the same base population. High body weight 1 (HBW1), low body weight (LBW), and layer (L) lines were selected for 15 generations according to 5-week high body weight, 5-week low body weight and 120 days egg yield, respectively. HBW2 line had been selected for 4-week high body weight for 7 generations. To see if different selection criteria and different generation numbers made differences on body weight, the selection studies of HBW2 was started after eight generations selection of other quail lines. Unselected control line was generated at same time the selection lines. The lines were established by applying individual selection with 10% and 40% selection intensity for males and females, respectively. Mating was random to minimize inbreeding.
A total of 69 individual birds from C (13), L (12), LBW (15), HBW1 (14), and HBW2 (15) representing five lines were used in this study. Approximately, 2 ml of blood was drawn from jugular vein into tubes treated with K3- ethylenediaminetetraacetic acid. Isolation of DNA from each line was performed using DNA Kit (Bio Basic Inc-SK252, Markham, ON, Canada). Each of the DNA samples was controlled by NanoDrop (ND 100) for quality and quantity. A set of fifteen SSR loci (GUJ0006_13_ 23_27_31_33_36_37_52_54_55_63_71_87_97) (Kayang et al., 2002) were chosen based on their degree of polymorphism for this study.
DNA amplification at polymerase chain reaction involved initial denaturation at 94°C (5 min) followed by 30 cycles of denaturation at 94°C (15 s), annealing at 55°C (15 s), extension at 72°C (15 s) and final extension at 72°C (20 min). A volume of 15 μL of reaction mixtures was prepared using 50 ng genomic DNA, 0.35 pmol/μL primer mix, 1× buffer, 25 μM dNTPs, 2.0 mM MgCl2, 0.04 U Taq DNA polymerase (Applied Biosystems, Waltham, MA, USA). Amplified DNAs were diluted by 1/10. Then, 12 μL formamide and 0.5 μL size marker (500 bp) were added to per 1 μL DNA sample taken from diluted samples. This mix was used to determine genotypes by automatic sequencer (ABI PRISMA 310, Lincoln Centre Drive, Foster City, CA, USA). GeneMapper 4.0 (Applied Biosystems, USA) was used to calling of SSR alleles.
POPGENE version 1.31 and CERVUS 3.0 (Kalinowski et al., 2007) package programs were used in the analysis of SSR genotypes data. Due to SSR markers showing co-dominant inheritance, a pattern counting method was used in the calculation of gene (allele) frequencies (Nei, 1987). Because allele number (na), effective allele number (ne), observed heterozygosity (Ho), expected heterozygosity (He), polymorphism information content (PIC), F-statistics and genetic distance (D) are accepted as the standard in SSR studies, these were used in determination of genetic variation of quail lines. The molecular phylogenic tree (unweighted pair group method with arithmetic mean [UPGMA]) analysis was performed to establish phylogenetic dendrogram. Genetic structure and the degree of admixture of quail lines were estimated using Bayesian clustering procedure of STRUCTURE 2.3 (Pritchard et al., 2000). First the program was run to assume the number of distinct populations defined as K. The analysis involved an admixture model with correlated allele frequencies. One hundred independent runs with 500,000 markov chain monte carlo iterations and a burn-in of 100,000 steps were performed for 2≤K≤8 (where K is the number of cluster to be tested) to estimate the most likely number of clusters present in the data set. The most plausible number of clusters was determined in the STRUCTURE HARVESTER (Earl and vonHoldt, 2012) by calculating the distribution of the ΔK statistic as described by Evanno et al. (2005). The clustering pattern was visualised using the software DISTRUCT 1.1 (Rosenberg, 2004).
RESULTS
All studied SSR loci were found to be polymorphic according to the results of the analysis over the quail genotypes used in this study. The lowest and highest Ho, He, and average heterozygosity (Ha) were estimated as 0.04 (GUJ0027) 0.64 (GUJ0087), 0.21 (GUJ0027), 0.84 (GUJ0037), and 0.20 (GUJ0027) 0.71 (GUJ0037), respectively (Supplementary Table S1, Supporting information). The na were determined as 3.47±1.06, 4.13±1.60, 4.00±1.20, 3.93±1.33, and 3.97±1.60 in C, L, LBW, HBW1, and HBW2, respectively, and the average of effective allele number (ne) was estimated as 3.26±1.28 (Table 1). The average observed heterozygosity over all loci in the C was 0.33±0.23 and found to be close to each other’s (L, LBW, HBW1, and HBW2; Table 1). In this current study the PIC values ranged from 0.18 (GUJ0063) to 0.69 (GUJ0037) (Supplementary Table S1) and the average PIC values was calculated from overall loci as 0.50±0.02 and ranged from 0.46±0.20 (HBW2) to 0.52±0.23 (L) (Table 1).
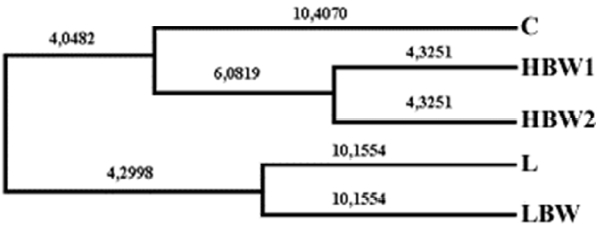
Average FST (genetic divergence of subpopulations within the total population) value was estimated as 0.13 and varied between 0.06 (GUJ0027) and 0.20 (GUJ0023). Values of coefficient of inbreeding (FIS) were between 0.03 (GUJ0087) to 0.81 (GUJ0031), with an average of 0.43. The inbreeding coefficient (FIT) average 0.50 and varied between 0.08 (GUJ0087) and 0.83 (GUJ31) in this study (Supplementary Table S1). FIS values ranged from −0.171 to 1.000 for the loci within the quail lines. The FIS values were calculated as relatively high positive and average Fis values varied between 0.382 (HBW2) and 0.460 (LBW). Hardy-Weinberg test over all Fis statistics demonstrated that the quail lines were not in equilibrium (Table 1). It can be understood that the homozygosity was increased due to selection pressure although there was random mating. Genetic distance values (Table 2) were determined from 0.09 (between HBW1 and HBW2) to 0.33 (between C and L). UPGMA cluster analysis was also shown as a dendrogram in Figure 1. Lines of C, HBW1, and HBW2 completed the first main cluster, L and LBW lines completed the second cluster in this dendrogram.
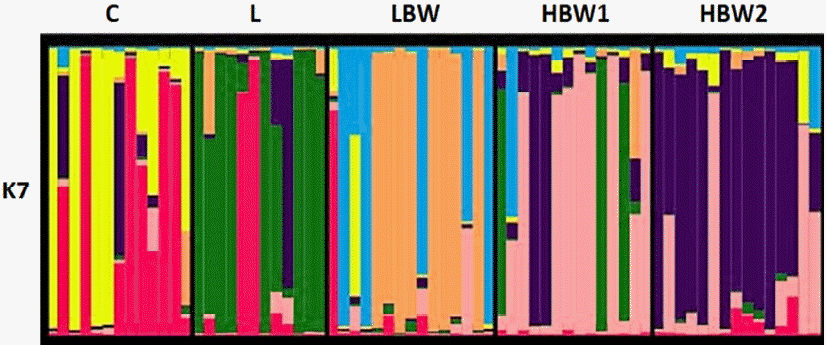
It can be said that the 5 Japanese quail lines were separated clearly based on 15 microsatellite markers in Figure 2. However, they could not fully differentiated at the end of 15 generations of selection. It was understood from common colors in Figure 2 that quail lines still carry the same genomic structures from a common ancestor.
DISCUSSION
When we examined the distribution of the alleles, we found that there were some differences in quail lines in terms of microsatellite alleles distribution. The average allele number over all loci was found to be 6.80±2.21 (Table 1). This value could be informative for such studies according to Farrag et al. (2011) who suggested that the average number of alleles per locus in the genetic studies distances must be >4 to reduce the standard error in the estimation of genetic distances. Average allele number found in studies that were carried on different quail genotypes by Chang et al. (2007), Farrag et al. (2011), Tadano et al. (2014) and Bai et al. (2013) as 2.33 to 5.67, 5.39, 5.20, and 5.3, respectively. As clearly seen here we found a higher allele number. The average of effective allele number (ne) was determined as 3.26±1.28 in this study (Table 1). When the distribution of alleles belonging to entire microsatellite loci in quail lines was examined, it appears that differential breeding of these quail lines for 15 generations has favored some alleles of the microsatellite loci of interest while not favoring others.
If the mean ne is high, which is defined as the inverse of homozygosity (Nei, 1987), it is more likely to detect the possibility of any genetic variation. Theoretically when all existing allele frequencies in a population are the same, ne should be equal to the value of na. But usually ne measure is estimated to be smaller than na. In this study, effective number of alleles belonging to the locus of lines was found to be between 1.27 (GUJ0027) and 6.09 (GUJ0037). The number of effective alleles were reported between 1.0 and 4.3 by Kayang et al. (2002), between 1.0 and 5.4 by Chang et al. (2007) and 4.03 by Bai et al. (2013).
When all of the working 15 microsatellite markers were analyzed, there were differences in expected and observed heterozygosity for some loci. It was found that expected and observed heterozygosity were lowest in loci GUJ002, but highest in GUJ0087 and GUJ0037 (Supplementary Table S1). Although the average observed heterozygosity values in all populations were lower than the average expected heterozygosity values (Table 1), there was not a significant difference between the lines in terms of the average observed heterozygosity values.
In a study performed by Chang et al. (2007) they reported higher heterozygosis values than reported here except for one microsatellite locus. Kayang et al. (2002) declared that observed, expected and average heterozygosis were as 0.00, 0.95, and 0.42 in Japanese quail, respectively. Also, the mean Ho was 0.609 and ranged across loci from 0.00 to 0.967 whereas the mean He was 0.636 and ranged between 0.139 and 0.802 in three Japanese quail strains (Farrag et al., 2011). Four Japanese quail strains were studied by using eight SSR loci and the expected heterozygosis varied between 0.708 and 0.849 (Amirinia et al., 2007). Ho and He per locus ranged from 0.00 to 0.54, and 0.12 to 0.80 (Tadano et al., 2014) and found as 0.63 and 0.60 (Gruszczynska and Michalska, 2013), respectively. Also, expected heterozygosity was estimated as 0.73 in wild quail and 0.23 in Japanese quail (Ahmad et al., 2014).
When PIC values were examined, it was seen that a substantial portion of working locus markers provided information at a high level. When Table 1 was analyzed in terms of PIC value, it was observed that there was a difference among quail lines. This value could be stated as higher than 0.477 (Kayang et al., 2002), but lower than 0.57 (Chang et al., 2007), 0.64 (Farrag et al., 2011), and 0.69 (Bai et al., 2013). Also, Tadano et al. (2014) reported that of 49 polymorphic microsatellite markers, 55.1% were highly informative (PIC≥0.50), 28.6% were reasonably informative (0.50>PIC>0.25) and 16.3% were slightly informative (PIC ≤0.25) according to Botstein et al. (1980) criteria. Gruszczynska and Michalska (2013) also found mean PIC value as 0.54.
In our study the value of FST was found as 0.13 within existing quail lines with respect to the microsatellite loci. According to definitions of Hartl and Clark (1997) this result can be interpreted as indicating a genetic differentiation among quail lines. There was 13% total genetic variation between quail lines and the remaining 87% genetic variation was due to differences within lines. FST was estimated for per locus from 0.22 to 0.92 and between each pair of lines ranged from 0.3 to 0.83 (Tadano et al., 2014). Values of FIS were between 0.03 (GUJ0087) and 0.81 (GUJ0031), the average of which was 0.43. FIS values were estimated 0.10 and 0.13 by Kim et al. (2007), 0.03 and 0.29 Chang et al. (2007). The inbreeding coefficient (FIT) was average 0.50 and varied between 0.08 (GUJ0087) and 0.83 (GUJ31) in this study. Kim et al. (2007) reported similar FIT as 0.57±0.33 in the 17th generation of inbred line using 14 microsatellite markers.
HBW1 and HBW2, which were selected in terms of higher body weight at 5 weeks for HBW1 and at 4 weeks for HBW2, were in the same cluster with some exceptions. The lowest genetic distance was found as 0.09 between HBW1 and HBW2 (Table 2). Chang et al. (2007) reported lower genetic distances (0.01 to 0.04) than found in this study (0.09 to 0.33). In another study, genetic diversity was ranged from 0.10 to 0.60 in 13 Japanese quail lines (Tadano et al., 2014).
The result of cluster analysis of 5 quail lines showed two main clusters (Figure 2). Cluster analysis largely realized our expectations regarding to genetic distance between quail lines. Namely, L line which were selected regarding to egg production was the most divergent one (Figure 2). HBW1 and HBW2 lines clustered on the same branch of the phylogenetic tree and these two lines can be evaluated as an indicator for the associated genes.
As expected, the number of clusters was found to be same as the number of the studied lines which was five as seen in Figure 2. Most of the individuals were visualized within their own clusters. However, it is clear that they still carry common genetic markers. Moreover, this genetic frame reflects what we found among the individuals genetic diversity. In a similar study, Tadano et al. (2014) demonstrated successful high genetic differentiation among 13 Japanese quail lines using Bayesian clustering (K = 13).
CONCLUSION
The selection made in different directions had separated the genotypes from each other to a certain extent. This result is consistent with the fact that the 5 quail lines studied here came from a common population and to obtain genetically unique lines selection needed to be applied for more generations and/or more markers must be used. However, this study would be helpful to support further selection studies using similar methodology. Also, we can say microsatellite markers can be effectively used for genetic characterization of selected quail lines.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







