



INTRODUCTION
Genomic selection (GS) has been substantially developed in the last few years (Usai et al., 2009; Ogutu et al., 2012), which estimates the total genetic value for animals utilizing the genomic information of a dense marker map covering all the chromosomes (Meuwissen et al., 2001). In practice, GS has been applied in an attempt to increase the accuracy of breeding values in various fields such as crop and livestock breeding (Iba├▒ez-Escriche and Gonzalez-Recio, 2011; Ogutu et al., 2012; W├╝rschum et al., 2013). For pig breeding, the different implementations of GS have been conducted to increase in accuracy of the breeding values (Simianer, 2009; Cleveland et al., 2010; Lillehammer et al., 2013). As interest in GS increased, the computational cost and the prediction accuracy of GS become important issues especially when the data are high-dimensional, i.e., the number of variables is larger than the number of observations.
Previously genome-wide association studies that utilize individual genes or a few quantitative trait loci (QTL) were popular (Meuwissen et al., 2001; Dekkers, 2002). However, it has a limitation in that it cannot reflect the effects of the neighborhood variables. In fact, the single nucleotide polymorphisms (SNPs) are ordered by physical locations on the chromosomes, therefore, adjacent SNPs are correlated with similar associations (Liu, 2011).
The main objective of this study is to provide significant SNPs that affect the average litter size of Yorkshire pigs using regularized regression approaches and to predict the litter values with selected SNPs in the final model. To accommodate the above property of SNPs properly, we consider some regularized regression approaches: least absolute shrinkage and selection operator (LASSO) (Tibshirani, 1996), fused LASSO (Tibshirani et al., 2005), and elastic net (Zou and Hastie, 2005). It is well known that these methods identify significant variables efficiently, improve the accuracy of the prediction and handle high-dimensional data simultaneously.
In fact, Ogutu et al. (2012) performed gnomic selection using some regularized regression methods. However, there are some key features that distinguish this study from Ogutu et al. (2012). First, in this study, we have analyzed ultrahigh-dimensional data that contain 519 sows with 47,112 SNPs, while Ogutu et al. (2012) considered a dataset that has 3,000 observations with 9,990 SNPs. It is well known that the performance of regularized regressions varies over the degree of dimensionality. Thus, this study can be considered as an extension of high-dimensional case of Ogutu et al. (2012). Second, an important issue of regularized regressions is their implementation when the data are high-dimensional, because it heavily involves solving optimization. To handle such ultrahigh-dimensional data, we have used three regularized regression methods with efficient algorithms introduced by Liu et al. (2010); and hence, many researchers can easily implement the methods for GS with various high-dimensional data. Finally, we have focused on the fussed LASSO. It is designed for a problem with features that can be ordered in some meaningful ways as well as a dataset where the number of features is much greater than the sample size. The abovementioned issues can be considered as main contributions of this study.
MATERIALS AND METHODS
The Rural Development Administration (RDA) provided the Illumina Porcine 60K SNP Beadchip on 703 sows and their litter size. Since this study is focused on the litter values of pigs, multiparous sows were used in the analyses. In this section, we explain the details of data in the analysis and the methods used for analysis.
Data
The original genotype data consisted of 60K SNP markers of 703 sows. Samples were excluded if they had a missing genotype rate (>0.05) per sample, and genotype data were also removed if they had low minor allele frequency (<0.01) or significant deviation from the Hardy-Weinberg equilibrium (p<0.0001) as determined by the Plink whole genome analysis toolset (Purcell et al., 2007). A quality control was performed when each SNP was recoded as having a value of 0, 1, or 2 for analysis.
There are several phenotype traits that genetically superior animals hold such as litter size by parity, gestation length, and number born alive. We considered the average litter size for sows as the response variable and the Illumina Porcine 60K SNPs as the independent variables. Although litter size is a trait with low heritability in pigs, it seems that, in our dataset, the litter size is the only response variable that could be used in regression models without imputation of feature variables (SNPs) and removing lots of pigs. The choice of response variable is not main issue of this study. As mentioned earlier, the main purpose of the paper is to compare the performance of three regularized regressions and to extract the influence SNPs for a particular trait when the data are ultrahigh-dimensional; and hence, it is feasible to use other traits that represent the productivity of pigs when such data are available.
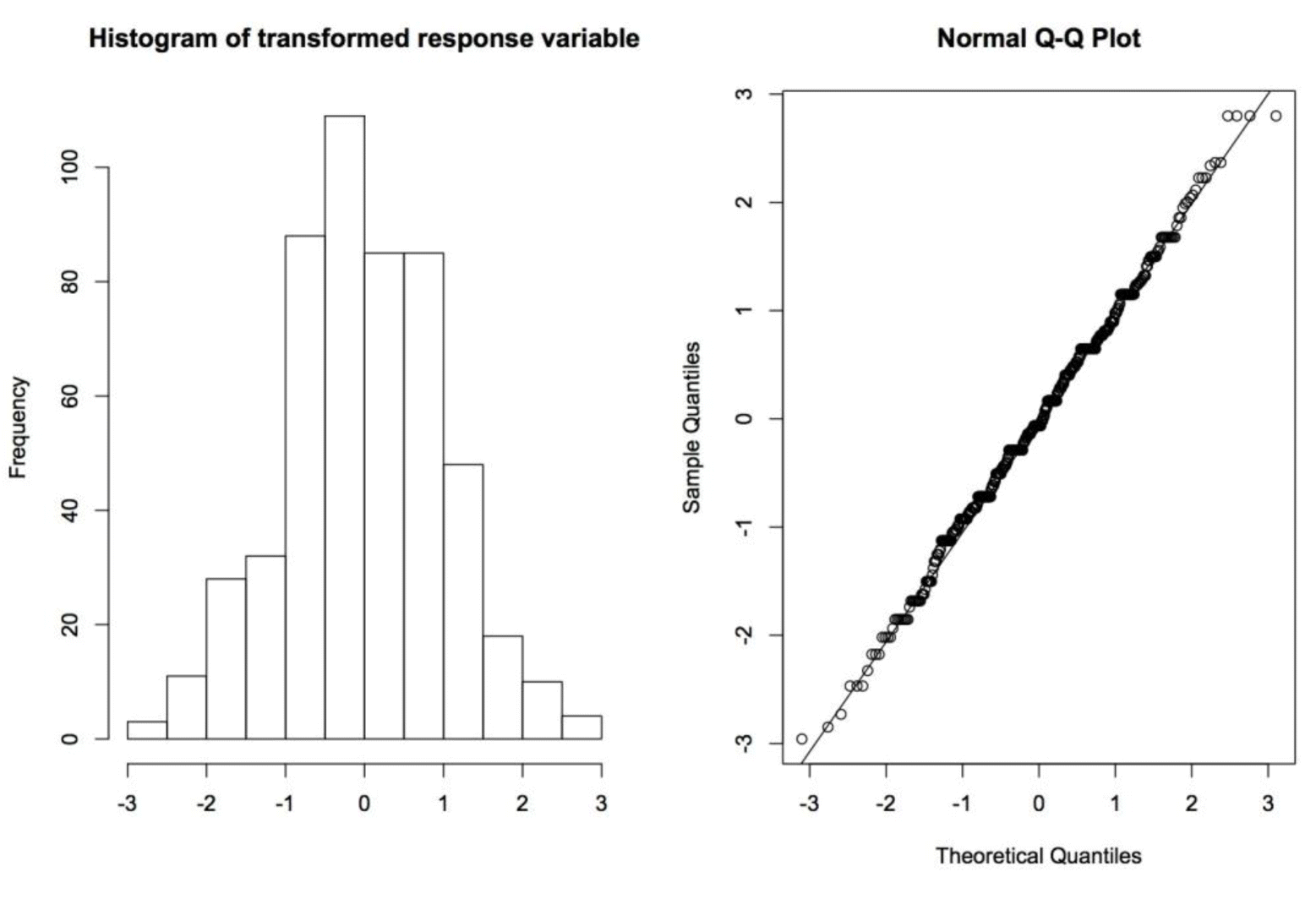
Litter size by parity in pigs is defined as the number of piglets born at a time and the parity of observed litter size in this data set ranges from 1 to 12. The observed objects per parity among a total of 4,163 pigs are described in Figure 1. The more the parity increases, the fewer objects there are (Figure 1). As shown in Figure 2, litter size per parity is almost identical as proved using the TukeyŌĆÖs honestly significant difference test. Therefore, in this analysis, we consider the average litter size for each sample matched with the objects of SNP marker information. The average values of litter size that can be addressed via the response variable must satisfy the Gaussianity assumption. To comply with this assumption, we used the Box-Cox transformation which chooses an optimal transformation to rectify deviations from the assumption. Figure 3 shows the empirical distribution of the transformed variable that satisfies the Gaussianity assumption. The Shapiro-Wilk normality test (Royston, 1982) gave a p-value of 0.3 with Shapiro-Wilk statistics of 0.9856. It implies that we cannot reject the Gaussianity assumption at the 0.05 significance level.
After phenotype and genotype realignment, the data set consists of 47,112 SNPs out of a total of 61,177 SNPs markers and 519 sows qualified for GS.
Figure┬Ā1
Observed sows per parity. The range of parity has from 1 to 12 and initial distinct observations having litter size values are 4,163 Yorkshire sows.


Methods
The linear model for the genetic effects at adjacent SNPs is
where y is an n├Ś1 vector of observed average litter size, X is an n├Śp matrix of genotypes, and ╬▓ is a p├Ś1 vector of the regression coefficients of the SNP markers. Here, ╔ø is an n├Ś1 vector of the i.i.d. random errors with
╔ø ~ N ( 0 , I Žā ╔ø 2 ) Žā ╔ø 2
LASSO (Tibshirani, 1996)
LASSO has been widely used for variable selection. It is used to find regression coefficients ╬▓ that minimizes the usual sum of squared errors with a constraint on the sum of the absolute values of the coefficients as follows
where the penalty function is P╬╗(╬▓) = ╬╗||╬▓||l1 with a regularization parameter ╬╗Ōēź0. Although LASSO is efficient and has a fast algorithm, it tends to arbitrarily select only one variable from the group when adjacent SNPs have pairwise high correlations, which may not be suitable for our analysis.
Fused LASSO (Tibshirani et al., 2005)
Fused LASSO is designed for a proper group selection that may be suitable for this study. The fused LASSO is defined as
As shown in the above criterion, the fused LASSO requires a natural ordering of the independent variables for integrating correlated variables well. The physical ordering of SNP markers across a chromosome is simply satisfied.
Elastic net (Zou and Hastie, 2005)
Elastic net is a regularized regression method that can be considered as an extension of LASSO. The penalty of elastic net consists of l1 and l2 parts, which means that it is a mixture of LASSO and ridge regression penalties. The quadratic penalty part captures the group of highly correlated variables, and hence, it overcomes a limitation of LASSO. The elastic net estimator can be expressed as
Letting ╬▒ = ╬╗2/(╬╗1+╬╗2), the estimator is equivalent to the following optimization problem:
subject to
( 1 - ╬▒ ) ŌĆ¢ ╬▓ ŌĆ¢ 1 + ╬▒ ŌĆ¢ ╬▓ ŌĆ¢ 2 2 Ōēż t
Comparing methods
Since the optimizations for LASSO, fused LASSO, and elastic net involve a non-linear procedure, finding the solutions is not trivial (Tibshirani and Taylor, 2011). To improve the speed and the accuracy of the solution paths, we used the efficient algorithm by Liu et al. (2010) with the accelerated gradient method of Nesterov (2007) and a two-deep 10-fold cross-validation (CV) in MATLAB as follows:
Partition the data into the first-deep training and validation sets; then partition the first-deep training set into the second-deep training and test set (two-deep CV).
At the second-deep, construct the model using the sub-training set and calculate CV; then choose the optimal tuning parameter that minimizes CV.
At the first-deep, fit the model and estimate the coefficients in the first-deep training set with the estimated regularization parameter from the second-deep set.
To evaluate the regularized regressions described in Section 2, we calculated the prediction error (PE), which is defined as
where the subindex val implies the validation set, β̂tra is a vector of the coefficients obtained from the first-deep training set and Xvalβ̂tra denotes a vector of the predicted values on the validation set. As a standard criterion for the performance, we consider mean fitting prediction error (MFPE) which replaces Xvalβ̂tra in the definition of PE with ȳtra.
RESULT AND DISCUSSION
To compare the performance of the three methods, PE and the Pearson correlation between the true litter values and the predicted values were used as measures of the accuracy. The number of non-zero coefficients was also used as an indicator of efficiency. If the structure of the training set is totally different from the arrangement of the validation set because of the limitation of data, then the evaluation of the method might not be reliable. To clarify the comparison for the results of methods, we extracted new samples from the original data using the bootstrap technique with a 35% resampling rate.
The overall results of each method with partitioned bootstrap samples are presented in Table 1 to 3. Table 1 describes PEs by LASSO, fused LASSO, and elastic net, and MFPE for each fold. The average PEs obtained by LASSO, fused LASSO, and elastic net were 0.8675, 0.8476, and 0.8628, respectively. As listed, the fused LASSO outperformed other methods when the explanatory variables were correlated. From Table 2, the fused LASSO provided the highest accuracy over all folds. Table 3 lists the number of non-zero coefficients selected by LASSO, fused LASSO, and elastic net. Overall, the number of non-zero coefficients by the fused LASSO was slightly high, compared to LASSO and elastic net. In the case of fold 4, for example, the consecutively selected variables by the fused LASSO were DRGA0005762, DRGA0005763, DRGA0005767, and DRGA0005770, while the LASSO selected only DRGA0005770. According to the results of the above comparison study, the fused LASSO performed well. Ogutu et al. (2012) analyzed a dataset with 9,900 SNP markers on 3,000 progenies of 20 sires and 200 dams by using various GS methods such as ridge regression, ridge regression best linear unbiased prediction (BLUP), LASSO, adaptive LASSO, elastic net, and adaptive elastic net. In their analysis, elastic net and LASSO worked well for GS, compared to others.
Table┬Ā1
Table┬Ā2
Accuracy (Pearson correlation1) and average correlation by regularized regression methods
| Fold | Regularized regression | ||
|---|---|---|---|
|
|
|||
| LASSO | Fused LASSO | Elastic net | |
| 1 | 0.3627 | 0.4150 | 0.3972 |
| 2 | 0.6802 | 0.6978 | 0.6966 |
| 3 | 0.6136 | 0.6410 | 0.6239 |
| 4 | 0.5600 | 0.5848 | 0.5694 |
| 5 | 0.7295 | 0.7510 | 0.7338 |
| 6 | 0.5973 | 0.6265 | 0.6011 |
| 7 | 0.4849 | 0.5126 | 0.4925 |
| 8 | 0.5931 | 0.6070 | 0.5962 |
| 9 | 0.5200 | 0.5422 | 0.5291 |
| 10 | 0.5708 | 0.5891 | 0.5777 |
| Ave corr1 | 0.5712 | 0.5967 | 0.5818 |
Table┬Ā3
Number of non-zero estimated coefficients derived from training set by regularized regression methods (total SNPs: 47,112)
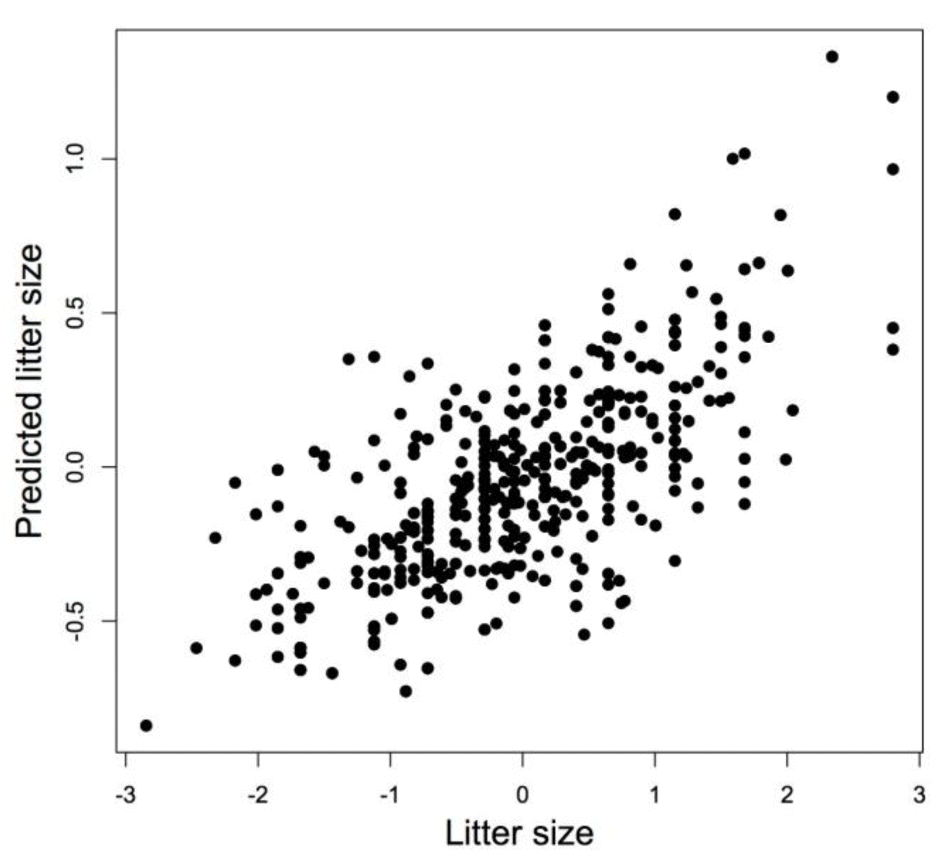
To construct the final model, we used the whole data set, and calculated PEs and correlation values between the given target variable and the predicted value to indicate accuracy. In the results of the final model obtained by the fused LASSO, the number of significant non-zero coefficients among 47,112 SNPs was 1,499 SNPs. Table 4 lists the names of the 10 selected significant SNPs with large estimated coefficients (in absolute value) of the SNP effects. Onteru et al. (2012) provided some important genes for reproductive traits in the QTL regions, where they employed the Bayes C model introduced by Kizilkaya et al. (2010). Figure 4 shows the scatter plot between the predicted values ┼Ę and the original values y. Note that the sample correlation coefficient between litter size and predicted litter size by the final model was 0.7041. It seems that the fused LASSO is suitable for selecting adjacent SNPs as well as for predicting the values.
In summary, we have considered three regularized regressions for GS with 47,112 SNPs of 519 sows. We compared three methods using PEs and correlation coefficient values, and obtained the final model to predict the litter size of pigs. From the data analysis, we observed that the fused LASSO seems to be a good choice for GS.
Figure┬Ā4
Scatter plot of true average litter size and predicted litter size obtained by the fused LASSO in the final model. The sample correlation coefficient is 0.7041. LASSO, least absolute shrinkage and selection operator.
Table┬Ā4
The 10 SNPs with the highest coefficients (in absolute value) selected by the fused LASSO1
| Name of SNP | Coef2 |
|---|---|
| M1GA0023299 | 0.0099 |
| MARC0015851 | 0.0094 |
| H3GA0002658 | 0.0084 |
| ASGA0001125 | 0.0074 |
| ALGA0106999 | 0.0069 |
| MARC0016306 | 0.0068 |
| ASGA0080059 | 0.0064 |
| ASGA0054467 | ŌłÆ0.0063 |
| MARC0027886 | ŌłÆ0.0064 |
| MARC0023564 | ŌłÆ0.0064 |
Finally, the regression methods for estimating the random effect in a mixed model have recently developed (Onteru et al., 2012; Resende et al., 2012). As a future study, it is worth confirming the effects of a random effect model compared to existing GS methods.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







