Survey of genetic structure of geese using novel microsatellite markers
Article information

Abstract
Objective
The aim of this study was to create a set of microsatellite markers with high polymorphism for the genetic monitoring and genetic structure analysis of local goose populations.
Methods
Novel microsatellite markers were isolated from the genomic DNA of white Roman geese using short tandem repeated probes. The DNA segments, including short tandem repeats, were tested for their variability among four populations of geese from the Changhua Animal Propagation Station (CAPS). The selected microsatellite markers could then be used to monitor genetic variability and study the genetic structures of geese from local geese farms.
Results
14 novel microsatellite loci were isolated. In addition to seven known loci, two multiplex sets were constructed for the detection of genetic variations in geese populations. The average of allele number, the effective number of alleles, the observed heterozygosity, the expected heterozygosity, and the polymorphism information content were 11.09, 5.145, 0.499, 0.745, and 0.705, respectively. The results of analysis of molecular variance and principal component analysis indicated a contracting white Roman cluster and a spreading Chinese cluster. In white Roman populations, the CAPS populations were depleted to roughly two clusters when K was set equal to 6 in the Bayesian cluster analysis. The founders of private farm populations had a similar genetic structure. Among the Chinese geese populations, the CAPS populations and private populations represented different clads of the phylogenetic tree and individuals from the private populations had uneven genetic characteristics according to various analyses.
Conclusion
Based on this study’s analyses, we suggest that the CAPS should institute a proper breeding strategy for white Roman geese to avoid further clustering. In addition, for preservation and stable quality, the Chinese geese in the CAPS and the aforementioned proper breeding scheme should be introduced to geese breeders.
INTRODUCTION
The output value of goose production is important to husbandry in Taiwan. At the end of 2014, there were 2.21×106 geese being raised on various farms in Taiwan and 5.55×106 geese that had already been slaughtered during that year [1]. There are two main breeds of geese in Taiwan, namely, Chinese geese and white Roman geese. Chinese geese were introduced from mainland China by early immigrants more than 300 years ago. They were divided into two lines, white and brown lines, by their coloring. Due to long-term separation and adaptation, the Chinese geese in Taiwan became differentiated from their counterparts in China [2]. There were no other goose breeds in Taiwan before the 1970s. However, in order to improve production efficiency, the Changhua Animal Propagation Station (CAPS) brought in white Roman geese from Demark in 1974 and from the USA in 1985. Equipped with a better growth rate and greater reproduction efficiency than Chinese geese, white Roman geese soon became the major geese population in Taiwan, accounting for 97% of the local geese market [3]. Meanwhile, the number of Chinese geese declined rapidly; hence, a subspecies conservation program was initiated at the CAPS in 1977. The CAPS is a breeding center for geese in Taiwan. The primary focus of the CAPS is on the conservation of various breeds, a goal that includes managing the breeds as appropriate for better utilization, with the aim of ensuring breed discrimination [2,4]. The significance of breeds lies not just in their separate identities; rather, each breed comprises a unique set of gene combinations, and as is now being increasingly recognized, each one has adaptive gene combinations [5]. Relatedly, information about genetic diversity and population structures is very important to draw the essential outline for any appropriate conservation and sustainable management program [6]. However, reports regarding the genetic diversity of domestic geese in Taiwan were relatively rare in the past, while the breeding programs utilized by geese farms were unpredictable. About fifteen years ago, Lin et al [7] elucidated genetic diversity among white Chinese geese, brown Chinese geese, and white Roman geese at the CAPS through sera protein and blood cell enzyme typing. For these three goose breeds, only four out of seven blood protein loci have polymorphisms. In terms of heterozygosity and the effective number of loci, the values for both types of Chinese geese were higher than those for white Roman geese. To date, however, the genetic information regarding domestic geese in Taiwan remains deficient. In 2015, multiple bird flu viruses were spread across and permeated many regions of Taiwan, resulting in massive mortality and slaughter among geese [8]. The scale of goose industry in Taiwan was decline temporarily and more geese were imported. The establishment of clear genetic information for goose populations is thus increasingly urgent in order to avoid breed loss and inbreeding. In the present study, we sought to use modern technology to analyze the genetic diversity and genetic structures of these poultry. Microsatellite markers are widely used for population genetics analysis of livestock. These markers contain abundant genetic information and can very clearly express the relationships between both individuals and populations. Moreover, they are commonly applied to assess diversity within breeds, inbreeding levels, breed differentiation, introgression, and breed admixtures [9].
White Roman geese, the European domestic breed, were de rived from grayleg goose (Anser anser), while Chinese geese, the Chinese domestic geese breed, were originated from swan goose (Anser cygnoides) according to analyses of their morphology and mitochondrial DNA polymorphism [10,11]. Microsatellite makers have already been applied in testing domestic geese for genetic diversity and genetic structures in some areas. In these efforts, due to the limited number of known markers, the investigators usually used markers which originated from different species or breeds. That caused many loci that have low allele numbers or unamplified polymerase chain reaction (PCR) [12–15]. Weiβ et al reported that several microsatellite markers were isolated from greyleg goose [16]. However, most of those markers revealed low polymorphism in our populations. In this study, new microsatellite loci were isolated, and these loci were then used to analyze the genetic diversity and genetic structures of the genetic resources in Taiwan.
MATERIALS AND METHODS
Experiment animals and sample collection
A total of 13 populations were investigated in this study. Five of these populations were from the CAPS, including white Roman geese (WR), white Chinese geese (WC), brown Chinese geese (BC), white Chinese and white Roman hybrid geese (H), and black swans (Cygnus atratus) (BS). Eight of the populations were from private farms, five of them being white Roman geese populations from five geese farms located in south and central Taiwan; these populations were identified as the Z, P, I, C, and Y populations, respectively. Another two of the eight private populations consisted of Chinese geese from farms located in Taoyuan (in the north of Taiwan) and Chiayi (in the south of Taiwan); these populations were identified as the TC and CC populations. The last of the aforementioned eight populations, a white Grimaud (WG) population, was made up of geese similar to white Roman geese in appearance, with the WG also having been introduced from Europe.
Blood samples were collected from 457 individuals from these 13 populations, including 99 WR, 40 WC, 63 BC, 36 H, 16 BS, 20 Z, 17 P, 18 I, 18 C, 16 Y, 39 CC, 35 TC, and 40 WG individuals. For each bird, 3 mL of blood were drawn from the superficial plantar metatarsal vein or wing vein. Whole genomic DNA was then extracted with Genomic DNA Isolation Reagent (GenePure Technology CO., LTD, Taichung, Taiwan) using the standard phenol-chloroform method.
Isolation of microsatellite loci from white Roman geese
Genomic DNA was isolated from blood taken from one male and one female white Roman goose using a previously described method; the following steps were slightly modified from the procedure previously described by Glenn and Schable [17]. The isolated genomic DNA was partially digested with RsaI and XmnI (NEB, Ipswich, MA, USA) until most of the DNA fragments were between 300 to 1,000 bp in length. Re-naturing of single strand SuperSNX24 forward (5′GTTTAAGGCCTAGCTAGCAGAATC 3′) and SuperSNX24+4P reverse (5′pGATTCTGCTAGCTAGG CCTTAAACAAAA3′) formed SuperSNX24 linkers that were ligated to the digested DNA fragments. Linked DNA fragments were then amplified with SuperSNX24. Next, microsatellite markers including fragments with biotinated probes were isolated and enriched. Three probe compositions were used: i) (TG)12, (ACT)12, (ACTG)6, and (ACAG)6; ii) (AG)12, (ACAT)8, (AACT)8, and (AAGT)8; and iii) (AAG)8, (AAAC)6, (AATC)6, and (AGAT)6. The biotinated probes then were annealed to fragments of gDNA containing complementary regions. Finally, the microsatellite-containing fragments were enriched using streptavidin-labeled metal beads (Dynabeads M-280 Streptavidin, catalog #11205D, Invitrogen, Carlsbad, CA, USA). The enriched segments were then cloned into pGEM-T Easy vector (pGEM-T Easy Vector system, Promega, Madison, WI, USA) and sequenced with an ABI 3730XL DNA Analyzer (ABI PRISM, Foster City, CA, USA). The fragments with higher repeats were then selected for polymorphism testing.
Polymerase chain reaction and polymorphism test
The selected highly repeated fragments were subjected to PCR and polymorphism testing which would verify if the microsatellite loci could be amplified and used to show diversity in the investigated populations. Primers for loci amplification were designed using Primer3plus [18]. CAG-tag (5′-CAGTCGGG CGTCAT CA-3′) or M13Reverse (5′-GGAAACAGCTATGACCAT-3′) was added to the 5′ end of one of each primer pair [19]. Following the protocol described by Schuelke [20], a fluorescent dye-labeled tag, as a third primer, was used with the primer pair to amplify the target fragments which were detectable on the capillary electrophoresis. Thirty unrelated white Roman geese from the CAPS were tested. PCR was performed on a 20 μL volume using a thermalcycler (GeneAmp PCR system 9700, Applied Biosystems, Foster City, CA, USA) containing 0.5 U Taq DNA polymerase (TAKARA, Kusatsu, Japan), 1×PCR buffer (1.5 mM MgCl2), 0.2 mM dNTP, 0.2 μM unlabeled primer, 0.04 μM tag-labeled primer, 0.16 μM dye-labeled tag, and 50 ng gDNA. The PCR cycling program was as follows: 95°C for 5 min, 35 cycles of 95°C for 30 s, 50°C to 65°C for 40 s, 72°C for 40 s, and a final elongation at 72°C for 7 min. The amplified microsatellite PCR products were analyzed with a DNA analyzer (ABI PRISM 3730 DNA analyzer, Applied Biosystem, USA). Allelic sizes of all loci were estimated relative to in-line GeneScan600 LIZ Size Standard marker (ABI PRISM, Applied Biosystem, USA). The fragment size was calibrated and analyzed with Peak Scanner Software version 1.0 (ABI PRISM, Applied Biosystem, USA). The loci which had an allele number greater than four and similar annealing temperatures were selected for whole population analysis.
Multiplex polymerase chain reaction
The 14 new and the seven previously known [16] microsatellite loci were selected (Table 1). The 14 new loci were included in one multiplex set and the seven known loci were included in another. Some primers were re-designed to ensure the proper length of PCR products to fit the multiplex demand, and the 5′ end of each forward primer was labeled with fluorescent dye (FAM, 6-fluorescein amidite; VIC, PET, or NED). Multiplex PCR was performed on a 30 mL reaction containing 1 U Taq DNA polymerase (TAKARA, Japan), 1×PCR buffer (1.5 mM MgCl2), 0.3 mM dNTP, 0.3 μM forward and reverse primers, and 100 ng DNA template. The steps of PCR program and genotype detection were the same as that described in the preceding subsection above except annealing temperature in multiplex 1 was 58°C and in multiplex 2 was 53°C.

Primer sequences, repeated motifs, fluorescent labeling and annealing temperatures of 21 microsatellite loci
Statistical analysis
For each locus and population and across populations, commonly derived statistics from the microsatellite genotypic data including allele frequencies, the observed number of alleles (No), the observed heterozygosity (HO), the expected heterozygosity (HE), and the polymorphic information content (PIC) were calculated with the Microsatellite Toolkit [21]. The Hardy-Weinberg equilibrium (HWE) test was performed using the GENEPOP computer program [22], which also was used to estimate F-statistics (FIT, FIS, and FST) [23] for each locus, the pairwise FST between populations, and the average inbreeding coefficient (FIS). Nei’s genetic distance (DA) [24] between populations was measured with Microsatellite Analyzer [25]. The phylogenetic tree was generated via the PHYLIP [26] program using the unweighted pair group method with arithmetic mean [27] and the neighbor-joining (NJ) method with bootstrap test of 1,000 resamplings of loci with replacement [28]. The genetic distances of the proportion of shared alleles (POSA) was used to estimate [29] and draw a POSA individual phylogenetic tree.
The model-based approach proposed for population structure analysis of the five CAPS populations was carried out with the software STRUCTURE 2.3.1 [30], which assessed the genomic clustering (K) of the sample. To obtain a representative value of K for data modeling, ten independent runs were performed for each value from one to seven. The run length was set to 100,000 burn-ins followed by 100,000 iterations. The ΔK estimated the most likely number of K that represented the population structure [31]. Subsequently, the CLUMPP software 1.1.2 [32] was used to demonstrate the optimal alignment of the 20 replicates for the same K value. The mean membership matrix across replicates was calculated with DISTRUCT v.1.1 [33]. Principal component analysis (PCA) was performed with GENALEX v.6.501 software [34,35] in order to spatially plot clusters and individuals based on the distance matrix with data standardization.
A hierarchical analysis of variance was carried out to allow the partitioning of total genetic variance into components owing to region, population, and individuals. Computations were carried out using a hierarchical analysis of molecular variance (AMOVA) procedure, as implemented in the ARLEQUIN 3.5 package [36].
RESULTS
Isolation of novel microsatellite loci and multiplex polymerase chain reaction
Over 200 DNA fragments containing short tandem repeats were cloned. A polymorphism test was performed using the CAPS populations, and 14 loci which had allele numbers higher than four were picked [37]. The 14 novel markers included one compound repeat (5A265141) and two complex repeats (5A26254 and 5A5279) [38]. In simple repeat markers, there was one hexanucleotide unit (5A5305-1), two pentanucleotide units (5A26648 and 5A26681), and one tetranucleotide unit (5A5397), while others contained dinucleotide units. These novel markers were then combined with the seven previously known markers, and together formed a set of markers for this study (Table 1). The multiplex PCR results are shown in Figure 1.

Electrophoretic diagrams of multiplex 1 and multiplex 2.
Polymorphism, heterozygosity, and F-statistics of selected microsatellite loci
Polymorphism was clearly observed at all the microsatellite loci in all five of the CAPS populations. The genetic characteristics of the 21 microsatellite loci are listed in Table 2. The average number of alleles per locus (Na) was 11.09. The actual number of alleles ranged from 5 (5A26261, 5A265164, Ans25, and Aph19b) to 38 (5A26681). The average number of effective alleles per locus (Ne) ranged from 1.678 (Ans17) to 18.48 (5A26681), with an average across loci of 11.09. The PIC value ranged from 0.369 (Ans17) to 0.943 (5A26681), with an overall average of 0.705. All of the selected microsatellite loci in this study were sufficiently polymorphic, indicating that these loci were suitable for the genetic analysis of geese.

Characteristics of 21 microsatellite markers used in 5 goose populations of CAPS
The H E among the 21 microsatellite loci had a range of 0.405 (Ans17) to 0.948 (5A26681), with the average value of HE being 0.745. The HO among the 21 microsatellite loci had a range of 0.083 (Ans13) to 0.897 (5A26648), with the average value of HO being 0.499 (Table 2). However, there were four loci, namely, 5A5111, 5A5305-1, 5A265175, and Ans02, that significantly departed from the HWE (p<0.01).
The Wright’s F-statistic values (FIS, FIT, and FST) for each locus are shown in Table 2. The average FIS for all the loci was 0.079, and the FIS per locus varied from 0.683 (Ans13) to −0.124 (5A265151). The average FIT for all the loci was 0.396, and the FIT per locus varied from 0.061 (5A26648) to 0.899 (Ans13). The mean FST for all the loci was 0.357. This value implied that around 35.7% of the total genetic variation was caused by population differences and that 64.3% of total genetic variation was due to genetic differentiation among individuals within each population.
Intra-population genetic variability and Hardy-Weinberg equilibrium test
The genetic statistics relating to polymorphism, including HE, HO, PIC, the mean Na, and the mean effective number of alleles, were calculated to estimate the allelic diversity at each locus of population. These genetic parameters across the 21 loci for the 13 goose populations are listed in Table 3. HE varied from 0.464 (WG) to 0.668 (H), whereas HO varied from 0.430 (WG) to 0.737 (H) and PIC ranged from 0.420 (WG) to 0.612 (H). The H population exhibited the highest values of HO, HE, and PIC, and the smallest value of FIS. The WG population had the lowest values of HO, HE, and PIC.

Genetic parameters across 21 loci in the 12 goose populations
Among the 13 populations, the CC population had the highest observed mean number of alleles (MNA) (7.000), followed by the H (6.200) and TC (5.900) populations, while the WC population had the smallest observed MNA (4.900). Negative FIS values were observed only in two populations (H and WR-I), indicating an insufficient degree of inbreeding. The deviation from the Hardy-Weinberg proportions within populations (FIS) varied from −0.101 to 0.205. The highest inbreeding effects were found in the TC population (0.205). The H population contained 16 loci that were significantly deviated from the HWE (p<0.01) (Table 3), and were much higher than other populations.
Inter-population genetic variation
To estimate genetic variation among the 13 goose populations, two parameters, genetic differentiation (FST) and genetic distance, were evaluated in this study. The values of genetic distance and genetic differentiation (FST) for each test population pair are summarized in Table 4. The FST for each population pair was highly significant (p<0.05), with exception of the private white Roman population pairs (Z-P, P-I, P-C, P-Y, I-C, and C-Y) and the population pairing of the C and WR populations. The BS population had the highest FST in relation to the other populations. The FST values of the remaining population pairs were varied from 0.036 (for the CC and TC population pair) to 0.492 (for the WG and TC population pair).

Pair-wise estimates of breed differentiation (FST) (below the diagonal) and genetic distance (above the diagonal) between each pair of the 13 goose populations
The genetic distances among the private white Roman pop ulations (that is, the Z, P, I, C, and Y populations) were fairly low (<0.1), while they were relatively high between the BS population and the other populations (>0.8). The genetic distances between the remaining population pairs varied from 0.098 (for the CC and TC population pair) to 0.703 (for the Z and TC population pair).
Population differentiation analysis
The Nei’s [39] genetic distance of these 13 populations of geese in Taiwan was also calculated. A DA distance matrix was used to build phylogenetic trees with the NJ method. A phylogenetic tree using the NJ method with bootstrap resampling (n = 1,000) of the 21 microsatellite loci was constructed with the PHYLIP software. In the NJ tree (Figure 2), the white Roman populations (WR, Z, Y, P, I, and C) and the Chinese goose populations (BC, WC, CC, and TC) were independently grouped into two clads. The Chinese populations were further split to two groups, one consisting of the BC and WC populations from the CAPS and another consisting of the CC and TC populations from private farms. The WG and H populations were depicted as independent taxa.

The unweighted pair group method was used with a neighbor joining (NJ) dendrogram summarizing genetic relationships among the thirteen goose populations based on Nei’s DA distances for the 21 microsatellite loci. The numbers on the nodes indicate the percentage bootstrap values generated from 1,000 resamplings. BC, brown Chinese geese; WC, white Chinese geese; CC, Chinese geese from private farm in Chiayi; TC, Chinese geese from private farm in Taoyuan; WG, white Grimaud geese; Z,Y,P,I,C, white Roman geese from Z,Y,P,I,C farms respectively; WR, white Roman geese; H, white Roman×white Chinese geese; BS: black swan (out-group).
A PCA of pair-wise genetic distances among the 13 examined goose populations was used to represent the relative positions of the populations. The first (PC1), second (PC2), and third (PC3) principal components accounted for 60.82%, 28.06%, and 3.27% of the total variation, respectively (Figure 3). The results of the PCA were similar to the phylogenetic tree drawn up via the NJ method except that WG was inseparable from white Roman groups.
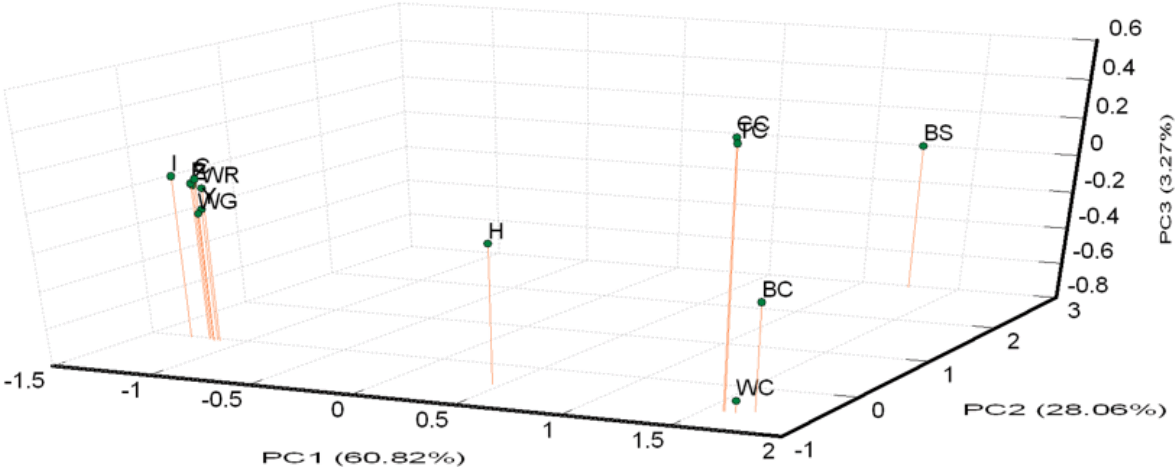
Principle coordinate analysis (PCA) plot of 13 population positions by population genetic distances based on the alleles frequencies of 21 microsatellite markers (loci). The first (PC1), second (PC2), and third (PC3) principal component accounted for 60.82%, 28.06%, and 3.27% of the total variation, respectively. BC, brown Chinese geese; WC, white Chinese geese; CC, Chinese geese from private farm in Chiayi; TC, Chinese geese from private farm in Taoyuan; WG, white Grimaud geese; Z,Y,P,I,C, white Roman geese from Z,Y,P,I,C farms respectively; WR, white Roman geese; H, white Roman×white Chinese geese; BS: black swan (out-group).
Population structure analysis
The degree of variance in the goose populations was quantified via the components AMOVA. The results are summarized in Table 5. In the analysis of all the populations, the largest variation was found within individuals (63%), followed by variation among populations (31%). The variation among individuals within populations accounted for 6% of the total variation. In the Chinese goose populations, the variation among individuals within the populations accounted for 15% of the total variation, while that among populations accounted for 9% of the total variation. If the Chinese geese were split to two populations, the variation among the populations was 9% in the private populations (the CC and TC populations) and 3% in the CAPS populations. In the white Roman populations, the variation within individuals reached 92% while that among populations was only 3%. Although the variation among populations in private Chinese goose populations was also 3%, the variation among individual Chinese geese was 18% in comparison to the 5% among white Roman geese.

Hierarchical analysis of molecular variance (AMOVA) within and among populations of goose
The STRUCTURE software program using Bayesian model- based clustering algorithms of multi-locus genotypes was utilized to assign individuals into populations via estimated individual admixture proportions and to infer the number of populations (K) for a given sample. The results of the analysis of all the populations are shown in Figure 4A. White Roman and Chinese geese were definitively separated into different clusters at K = 2, while black swans became an independent group at K = 3. At K = 5, the WG population was split into a new cluster. No new cluster appeared, however, and the figures were not much different for values of K larger than 6. In order to survey the structures on and intra-breed basis accurately, analyses of the white Roman and Chinese geese, respectively, were carried out (Figure 4B, 4C). Among the white Roman geese, the CAPS population comprised two subgroups at K = 2, and subsequently the white Grimauds became a new subpopulation at K = 3. The private populations always congregated in one subgroup until K reached 4. Among the Chinese geese, the CAPS and private populations could be divided into two clusters roughly at K = 2. However, white Chinese geese became a new whole group at K = 3, while the two private populations still stayed in one group. To investigate the private Chinese geese populations in detail, a STRUCTURE analysis for only two of the populations was performed (Figure 4D). At K = 2 and K = 3, the two populations included two and three components, respectively, with different proportions. At K = 4, the two populations finally had their own components. Most of the individuals in these two populations contained complicated components and were hard to define in terms of their grouping inclination.
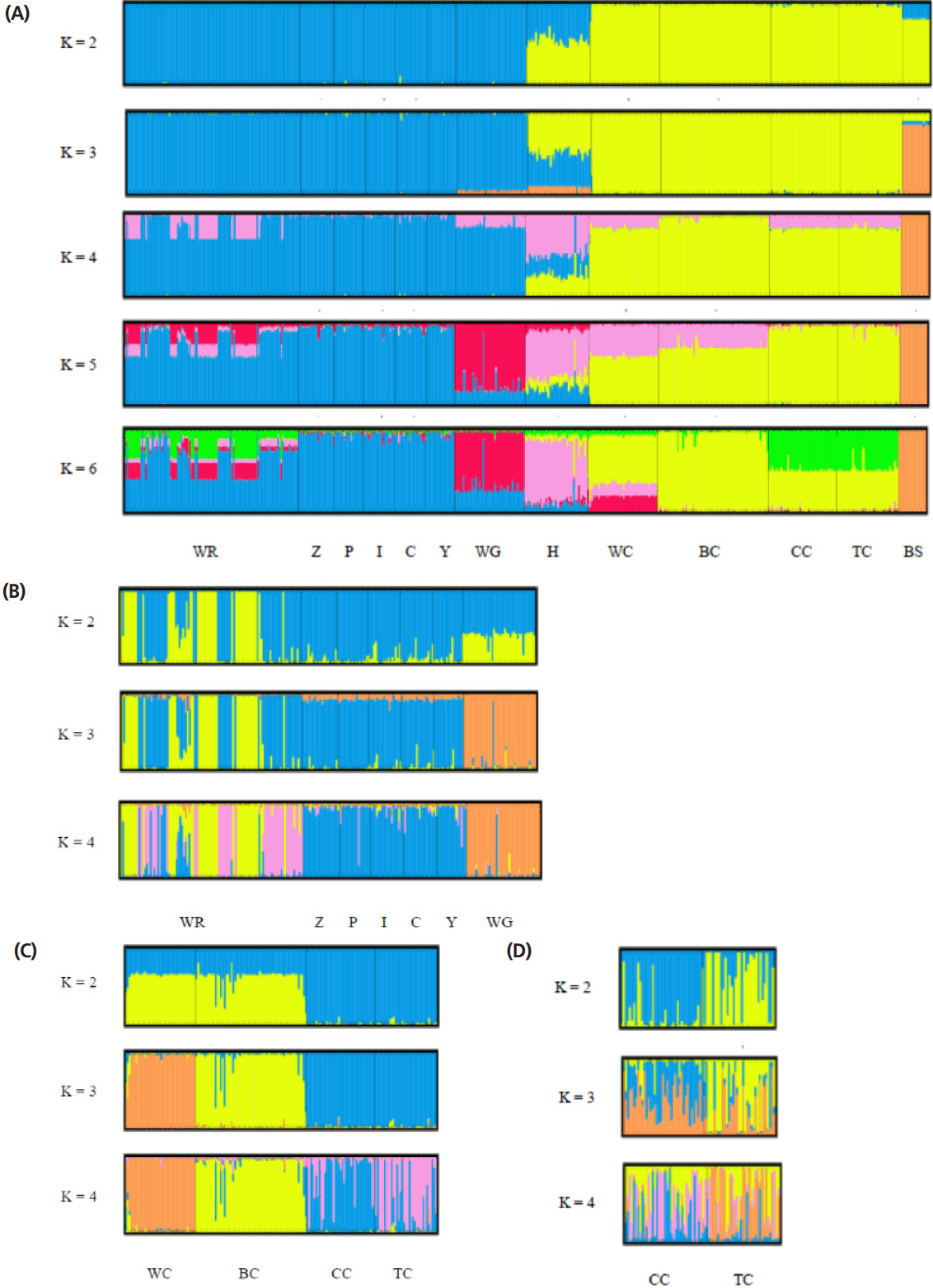
Structural analyses of goose populations. Each genotyped rabbit is represented by a single vertical line divided into K colors, where K is the number of clusters assumed in each structure analysis. Each vertical bar represents an individual rabbit. The colors on a vertical bar represent the probability that an individual belongs to that cluster. (A) Cluster results from a structural analysis of 457 geese from 13 populations (based on 21 microsatellite markers). Even though K>6, there was no new cluster appearance and the figures were not too different. (B) Clustering analyses of white Roman and white Grimond geese. (C) Clustering analyses of Chinese goose populations. (D) Clustering analyses of private Chinese goose populations. WR, white Roman geese; Z,Y,P,I,C, white Roman geese from Z,Y,P,I,C farms respectively; WG, white Grimaud geese; H, white Roman×white Chinese geese; WC, white Chinese geese; BC, brown Chinese geese; CC, Chinese geese from private farm in Chiayi; TC, Chinese geese from private farm in Taoyuan; BS: black swan (out-group).
DISCUSSION
There were 14 novel microsatellite loci that were isolated in this study. Among these loci, a number of hypervariable loci with more than 30 alleles in the CAPS populations, including 5A5279, 5A26648, and 5A26681, were found. Such complex and hypervariable loci have previously been found in human CODIS loci and some canine microsatellite loci [40,41] as well. These complex loci might be beneficial for individual differentiation due to their excellent diversity. The probability of identity values [42] of 5A5279, 5A26648, and 5A26681 were 0.13, 0.028, and 0.019, respectively in the white Roman CAPS population, and their combined value was 6.91×10−5, which means these three markers alone could be used to differentiate more than ten thousand individuals. In addition to high number of repeat units, the partial repeats and insertion-deletion in the flanks of repeat regions, one motif of 5A26648 was TAGAA (GAGAA)34 and another was TAGCAGAA (GAGAA)35, for example, caused variant alleles and broad allele range. Researchers should thus be careful about misreading it when performing electrophoresis and multiplex design.
To examine the diversity of the 14 novel microsatellite makers and the seven published markers in Taiwan geese, several indicators were calculated from randomly selected samples from the CAPS. The Ho values of all the markers were higher than 0.3, with the exception of Ans13 and Aalμ1b. A previous report suggested that microsatellite markers used in studies of genetic variation and distance should have HO values of between 0.3 and 0.8 in the population [43]. The Na values in this study always had at least five alleles (ranging from 5 to 38 alleles) in order to reduce the standard errors of the distance estimates [36]. The Na, Ne, and PIC values across all the loci were 11.09, 5.145, and 0.705, respectively, indicating higher genetic variability and diversity in the investigated geese than among domestic geese from other areas [12,14,44]. Significant deviation from the HWE was observed in only four of the 21 loci. An un-rooted individual phylogenetic tree (Figure 4) showed that most of the individuals had been sorted to their population distinctly and that the H population was located just between the white Roman and Chinese geese. These results indicated that the compilation of all the microsatellite markers used in this study were suitable for evaluating the genetic structures of goose populations in Taiwan.
Intra-population genetic variation and diversity
Table 3 presents the intra-population genetic variation in 12 of the geese populations. Deficiencies of heterozygosity (that is, HO lower than HE) were exhibited by almost all the populations. Only the H population had excess heterozygosity, while the WR and I populations each had roughly equal HO and HE values. The first generation of white Roman crossed Chinese geese have some characters: the excess heterozygosity (HE = 0.668, HO = 0.737), far from inbreeding (FIS = −0.101) and departure HWE (16 of 21 markers HWE departure). The white Roman geese from the CAPS had deficient heterozygosity and seven markers that departed from the HWE, but its FIS was nearly zero. With STRUCTURE test (Figure 3B), subpopulations were branched out in this population. This was probably caused by selection. For the five private white Roman farm populations, the levels of heterozygosity were deficient, the average FIS was 0.063, and the numbers of loci departing from the HWE were less than 2. It can thus be concluded that the white Roman farms in Taiwan probably have good breeding programs. The results of the intra-population diversity analyses for the two Chinese populations from the CAPS were similar, including relatively low variation and FIS values of around 0.15. This was probably caused by the populations being both closed and small for long periods of time. The CC population had a high MNA, medium high FIS (0.144), and seven markers that departed from the HWE. It was thus speculated that new genes were imported into the population relatively recently. The genetic diversity of the TC population was similar to that of the Chinese population from the CAPS except for the former’s high FIS (0.205). The TC farm has a serious inbreeding problem and urgently needs to improve their breeding program.
Inter-population genetic diversity and relationships
White Roman and Chinese geese are the two major domestic breeds in Taiwan. According to the AMOVA results, the proportion of genetic variation attributed to population differences in this study was about 28%, while the proportion of genetic variation attributed to individuals within populations was 6% of the total genetic variation. However, the individuals within populations were comparatively homogeneous, and phenomenon which might have been caused by the large differences between the European and Asian geese. This hypothesis was confirmed with the drawing of the NJ phylogenetic tree, which showed two major clads of Chinese and European (including white Grimaud) geese (Figure 2), and through the STRUCTURE analysis, which revealed that the European, Chinese, and BS populations were divided into three parts when K = 3, with the H population split into two contributions (Figure 4A). The PCA plot showed that the European and Chinese geese were significantly separated in terms of PC1, while the hybrid geese were located between them (Figure 3). The individual phylogenetic tree (Figure 4) also divided the Chinese and European geese into two clusters, with the hybrid geese inserted between them. The hybrids in the CAPS were created in a trial of two-way crossings for the improvement of carcass characteristics [2]. In our distinct analyses, the H population was always situated between two major groups or bisected individuals at K = 2 and K = 3. The 21 microsatellite markers used in this study have the ability to detect any crosses between European and Asian geese.
The samples of white Roman geese in this study were from the CAPS and five private farms. The proportion of genetic variation that came from population differences was only 3%. The six populations were hard to separate in 3D PCA and in the individual phylogenetic tree. The selected white Roman geese were rarely differentiated in terms of population genetics. Furthermore, when the CAPS populations were excluded, the among-population difference dropped to 1%. The inter-population variations of the CAPS populations were thus slightly higher than those of the five private farm populations. The white Roman geese from the CAPS were even split into two clusters of European geese in the STRUCTURE analysis at K = 2 (Figure 4B). Selection for growth and reproduction traits in a closed population may have been the cause of this [45]. In contrast, the five private populations remained in one cluster even at K = 4. Because of the highly homologous genetic structures among breeding geese, the genetic resources of white Roman geese in Taiwan were consistent and stable. The white Grimaud goose, a commercial breed that is similar to the white Roman goose in appearance, was also introduced from Europe. White Grimaud geese are rare in Taiwan, but they have possibly crossed with and influenced the white Roman breed. Genetically, the white Roman and white Grimaud populations in this study were hard to separate in the PCA (Figure 3). However, the white Grimaud geese could form an independent group without mixing with the white Roman populations in the individual phylogenetic tree (Figure 5) and formed a divided cluster at K = 3 in the European goose STRUCTURE analysis (Figure 4B). Based on these results, the white Roman populations do not seem to have been disturbed by white Grimaud geese.

The unrooted individual phylogenetic tree of thirteen geese populations constructed from –ln (shared allele proportion) by 21 microsatellite markers polymorphism.
 : WR,
: WR,
 : WC,
: WC,
 : BC,
: BC,
 : BS,
: BS,
 : H,
: H,
 : Z,
: Z,
 : Y,
: Y,
 : P,
: P,
 : I,
: I,
 : C,
: C,
 : WG,
: WG,
 : CC, ■: TC. WR, white Roman geese; WC, white Chinese geese; BC, brown Chinese geese; BS: black swan (out-group); H, white Roman×white Chinese geese; Z,Y,P,I,C, white Roman geese from Z,Y,P,I,C farms respectively; WG, white Grimaud geese; CC, Chinese geese from private farm in Chiayi; TC, Chinese geese from private farm in Taoyuan.
: CC, ■: TC. WR, white Roman geese; WC, white Chinese geese; BC, brown Chinese geese; BS: black swan (out-group); H, white Roman×white Chinese geese; Z,Y,P,I,C, white Roman geese from Z,Y,P,I,C farms respectively; WG, white Grimaud geese; CC, Chinese geese from private farm in Chiayi; TC, Chinese geese from private farm in Taoyuan.
The Chinese goose is not currently a major breed in Taiwan, but it does have a long history in Taiwan. The major purpose of raising Chinese geese in the CAPS was genetic source conservation for both white and brown Chinese geese, which generally were considered as a breed standard in Taiwan. We used several tools to analyze if the genetic structures of the private goose populations were distinct from the standard populations. In AMOVA, the proportion of genetic variation attributed to population differences in the CAPS Chinese goose populations was about 9%, which was more than the 3% proportion of variation in the two private Chinese populations. The two private Chinese geese populations also did not separate from each other but did separate from the CAPS Chinese geese in the 3D PCA. The FST value (Table 4) of CC vs TC was 0.036, the smallest among the Chinese geese populations, including the 0.088, 0.121, 0.118, 0.089, and 0.095 values of WC vs BC, WC vs CC, WC vs TC, BC vs CC, and BC vs TC, respectively. The white and brown geese from the CAPS were obviously different in terms of their genetics, but also the differentiability of the private and CAPS Chinese geese was greater than that of the white and brown Chinese geese in the CAPS. Although two private populations were similar in the PCA, the details indicated by the STRUCTURE analysis could effectively rule out the previous analysis. The results of the STRUCTURE analysis of the two private populations (CC and CT) showed that they have different proportions of two portions at K = 2 (Figure 4D). Meanwhile, at K = 4, the differences were enhanced, such that the CC population had a greater pink portion and the TC population had a greater orange portion. Moreover, the intra-population variation of in the two private farm populations was high according to the AMOVA and STRUCTURE analyses. The inter- and intra-population differentiations were probably caused by migration and crossbreeding with other breeds.
CONCLUSION
The present study verified that the 21 microsatellite loci, including the 14 novel loci isolated in this study, were useful in studying the relationships and genetic diversities among the goose populations in Taiwan. In addition, these markers could be applied in genetic content monitoring for quality analysis of geese. After an analysis involving 21 markers, several industrial white Roman geese farms revealed unified genetic structures in their breeders. The management of white Roman genetic resources in Taiwan thus seems reliable in terms of providing stable qualities. However, continuous selections performed in the CAPS have caused clustering within populations. The individuals were selected as seed from two subpopulations to mate each other. Thus, more stable and better performing populations could be produced. On the other hand, the white and brown Chinese geese raised in the CAPS became two genetically unique and unified populations via breed conservation efforts undertaken for about 30 years. However, Chinese geese raised at private farms revealed an uneven structure and were distinct from the CAPS populations. These results indicate that breeding management requires urgent improvement. The CAPS could provide their breeders for the geese industry in Taiwan in order to ensure stable production, maintain regional genetic resources, and develop hybrid geese for better meat quality.
ACKNOWLEDGMENTS
This work was financially supported by the Council of Agriculture, Executive Yuan, Taiwan (Grant No.: 100 AS-5.3.1-ST-aD and 101AS-6.1.1-ST-aC) in Taiwan. We would like to acknowledge the National Center for Genome Medicine, National Science Council, Taiwan, for their technical support.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.