Association of the Single Nucleotide Polymorphisms in RUNX1, DYRK1A, and KCNJ15 with Blood Related Traits in Pigs
Article information
Abstract
The aim of this study was to detect positional candidate genes located within the support interval (SI) regions based on the results of red blood cell, mean corpuscular volume (MCV), and mean corpuscular hemoglobin quantitative trait locus (QTL) in Sus scrofa chromosome 13, and to verify the correlation between specific single-nucleotide polymorphisms (SNPs) located in the exonic region of the positional candidate gene and the three genetic traits. The flanking markers of the three QTL SI regions are SW38 and S0215. Within the QTL SI regions, 44 genes were located, and runt-related transcription factor 1, dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A), and potassium inwardly-rectifying channel, subfamily J, member 15 KCNJ15–which are reported to be related to the hematological traits and clinical features of Down syndrome–were selected as positional candidate genes. The ten SNPs located in the exonic region of the three genes were detected by next generation sequencing. A total of 1,232 pigs of an F2 resource population between Landrace and Korean native pigs were genotyped. To investigate the effects of the three genes on each genotype, a mixed-effect model which is the considering family structure model was used to evaluate the associations between the SNPs and three genetic traits in the F2 intercross population. Among them, the MCV level was highly significant (nominal p = 9.8×10−9) in association with the DYRK1A-SNP1 (c.2989 G<A), and explained 4.9% of the phenotypic variance. However, since linkage disequilibrium is quite extensive in an F2 intercross, our approach has limited power to distinguish one particular positional candidate gene from a QTL region.
INTRODUCTION
Candidate genes generally refer to the genes that are known to have biochemical and physiological functions that can influence the variation of specific genetic traits. The effects of genetic variations within genes on phenotypic variations are generally verified through an association analysis. While a number of candidate genes have repeatedly been identified regarding economic traits, in particular, the quality of meat and the amount of meat, the number of genes that are applied to the actual industry after official acceptance―such as in the improvement and selection of livestock―remains small. “Candidate gene analysis” is an essential phase that is performed after quantitative trait locus (QTL) mapping to detect major variations of genetic traits. Linkage studies and candidate gene analysis on seemingly influential chromosomal regions are simple methods used to find candidate genes. The combination of the two methods can be more effective in finding candidate genes with higher accuracy (Lou et al., 2006; Zhu and Zhao, 2007). However, over 100 genes are generally included in the logarithm of odds (LOD) support interval (SI) for QTL. Therefore, even if high-density markers are used, more than 10 genes are still positioned (Ron and Weller, 2007). In this regard, the identification of candidate genes by taking into account the various genes located in target regions (QTL SI regions) has the disadvantages of being time consuming and expensive.
In our previous study, we detected highly significant QTL affecting the mean corpuscular volume (MCV) level and suggested QTL for red blood cell (RBC) and mean corpuscular hemoglobin (MCH) levels in Sus scrofa chromosome 13 (SSC13). In addition, we proposed that the runt-related transcription factor 1 (RUNX1) gene, which plays a major role in the development of blood cells (erythroid cells, granulocytes, macrophages, and lymphoid cells), may be a positional candidate gene for these genetic traits (Cho et al., 2011). In this study, the aim was to detect the positional candidate gene for RBC, MCV, and MCH QTLs in SSC13 and to examine the association of the positional candidate gene with the three genetic traits in F2 intercross between Landrace and Korean native pigs (KNP).
MATERIALS AND METHODS
Experimental animals and phenotypic analysis
A three-generation resource population was generated and managed as described by Cho et al. (2011). Briefly, nineteen purebred KNP were crossed with seventeen purebred Landrace. They produced 91 F1 progeny and 1,105 F2 progeny (568 males and 537 females) from 79 full-sib families. All experimental breeding and feeding took place in the same houses on Jeju Island as described by Cho et al. (2011). A total of 816 animals of the F2 generation were used for blood parameter analysis. Blood samples of 10 mL were collected at 140 days and were measured using a Scil Vet abc haematology analyzer (Scil Animal Care Company GmbH, Viernheim, Hesse, Germany). In the F2 generation, the phenotypic correlations between traits were measured using Pearson’s correlation test (Table 1).

Statistics of RBC, MCV, and MCH level from the F2 intercross between Landrace and Korean native pigs
List of positional candidate genes and polymorphism detection
Positional candidate genes within the QTL region for RBC, MCV, and MCH were searched on the NCBI database (http://www.ncbi.nlm.nih.gov). The scope of the search was between SW38 and S0215, and information about the genes located between the two flanking markers was collected and the positional candidate genes were selected according to their functions. To detect parental-specific single-nucleotide polymorphisms (SNPs) within the positional candidate genes, genomic DNA samples were selected from each of the two parental lines (KNP [n = 5] and Landrace [n = 5]), and the final concentration was pooled at 200 ng/μL. A massively parallel sequencing technology was applied to identify the SNPs in the entire porcine genome using pools of parental genomic DNA samples (HiSeq2000, Illumina Inc., San Diego, CA, USA).
Genotype analysis
The genotypes of SNPs were analyzed using the PCR (polymerase chain reaction)–RFLP (restriction fragment length polymorphism) method and the pyrosequencing method (Biotage AB, Uppsala, Uppland, Sweden). PCR was carried out in a total volume of 25 μL containing 50 ng DNA, 10× reaction buffer (with 20 mM MgCl2), 10 pmol each primer, 20 mM dNTPs, and 0.1 units Taq polymerase (GenetBio, Daejeon, Korea). A five minute denaturation step at 95°C was followed by 40 cycles of 30 s at 94°C, 30 s at the appropriate annealing temperature, and 30 s at 72°C, followed by a final extension of 5 min at 72°C in a PTC-200 Programmable Thermal Controller (MJ Research, Inc., Waltham, MA, USA). Four μL of each PCR product was digested overnight with 8 unit specific restriction enzymes at optimal reaction temperature. The digested PCR products were then separated on 4% agarose gel stained with ethidium bromide.
Statistical analysis
Similar to the QTL analysis of the previous study, the genotype of the analyzed SNPs was assumed to be diallelic and the alleles that could be selected from the two parental breeds were assumed to be “QQ” (with effect a) inherited from KNP and “qq” (with effect -a) inherited from Landrace. Prior to association analysis, the genotypes, alleles, and minor allele frequencies (MAFs) were evaluated. MAF<0.05 were excluded from the association analysis using a mixed model.
To measure the additive and dominance effects of the SNPs, additive and dominance components were produced. The additive components were set as constants according to the analyzed genotypes: 1 for QQ, 0 for Qq (or qQ), and −1 for qq. The dominance components were set at 0 for QQ and qq, and 1 for Qq (or qQ). The genetic effects and statistical significance of each component produced were confirmed using the Minitab program (Minitab Inc., State College, PA, USA) (p<0.05). The association between the phenotypes and genotypes of the SNPs was verified using the SAS software program, considering the effect of family. The following model was used to detect the significance of SNP:
Where, Yijklm is the phenotype of the ith F2 offspring of the jth batch from the cross of the kth sire and lth dam, μ is the mean value, sexi, and batchj are the fixed effect. Also, Sirek and Daml(k) are the random effects of the family, X is the weight at days of 140 as covariate, SNPm is the SNP genotype effect of the mth F2 offspring, ɛijklm is the residual error. The reduced model excluded the SNP genotype effects (fixed effects), from the full model. Means are reported as least squares mean±standard error. Differences were considered significant at p<0.05. The phenotypic variance (% Var) that was explained by a SNP genotype was obtained by the following equation using the residuals of the full and reduced models:
Where, RSSr is the residual sum of squares of the reduced model and RSSf is the residual sum of squares of the full model.
RESULTS AND DISCUSSION
List of candidate genes within the QTL SI region
The QTL regions for the RBC, MCV, and MCH levels were compared with the orthologous regions on human chromosomes to detect the positional candidate genes associated with the traits. In human chromosome (HSA) 2, 3, 18, 21, and Y were orthologous with SSC13; among them, q-arm of HSA21 was orthologous with the QTL region in SSC13. The trisomy of HSA21 is the cause of Down syndrome, which is a hereditary disease that causes mental retardation, physical deformation, functional disorder of the whole body, and growth impairment. The clinical disorders exhibited in Down syndrome are known to occur as numerous copies of the genes located in HSA21 induce the overexpression of genes (Mao et al., 2005; Leshin, 2006). Among the various clinical characteristics of Down syndrome, in terms of hematological values, this disorder is characterized by high levels of MCV, mean platelet volume, and neutrophil alkaline phosphatase (de Alarcon et al., 1984). Among these, a number of previous studies have reported that erythrocyte macrocytosis, which refers to a high level of MCV, was exhibited in the disorder (Wachtel and Pueschel, 1991; David et al., 1996; Kivivuori et al., 1996; Dixon et al., 2010; Tenenbaum et al., 2011).
The SI regions of RBC, MCV, and MCH QTLs in SSC13, which had been detected in the previous study, were observed to range from 95 to 106 cM, based on the 1.5-LOD drop method (Cho et al., 2011). Their flanking MS markers were SW38 and S0215. The physical locations of SW38 and S0215 were identified on the NCBI database, and the information on the genes located between the two MS markers was collected. As a result, it was confirmed that 44 genes existed between the flanking markers (Supplementary Table S1). Among them, we chose three genes, which are RUNX1, dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A), and potassium inwardly-rectifying channel, subfamily J, member 15 (KCNJ15), based on their reported functions.
Runt-related transcription factor 1 (RUNX1, NM_001246252.1), which is also known as acute myeloid leukemia 1 (AML1), encodes a runt-related transcription factor. In fact, it was suggested as a positional candidate gene in our previous study. In humans, the translocation between the RUNX1 gene on HSA21 and the eight-twenty-one (ETO) gene on HSA8 is known to be associated with acute myeloid leukemia (Asou, 2003). RUNX1 is also reported to play a major role in the control over the differentiation of hematopoietic stem cells in mature blood cells and the development of all blood cells (erythroid cells, granulocytes, macrophages, and lymphoid cells) (Okuda et al., 2001, Adamo et al., 2009). Moreover, in terms of the development of organs and tumors, RUNX1 is known to play an important role in hematopoiesis, neural development, and leukemogenesis (Speck and Gilliland, 2002; Yoshikawa et al., 2007).
Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A, XM_003132770.4) is known as participates in various cellular processes and encodes a member of dual-specificity tyrosine phosphorylation-regulated kinases. In the human, DYRK1A is located in chromosome 21q22.13, which is called the Down syndrome critical region. Shindoh et al. (1996) suggested that the human DYRK1A protein may play a significant role in a signaling pathway regulating cell proliferation and may be involved in normal brain development and in the pathogenesis of Down syndrome. In addition, the haploinsufficiency of DYRK1A in the development of the human brain has been reported to cause unique clinical symptoms such as mental retardation, primary microcephaly, intrauterine growth retardation, facial dysmorphism, motor dysfunction, and behavioral disorders, which are the characteristics of Down syndrome (Altafaj et al., 2001; van Bon et al., 2011). We suggested that the DYRK1A gene would also play an important role in the MCV level of pigs based on the finding that erythrocyte macrocytosis was exhibited in the clinical features of Down syndrome cases in previous studies.
Potassium inwardly-rectifying channel, subfamily J, member 15 (KCNJ15, XM_00005670338.2) encodes potassium channel inward rectifiers (Kir) 4.2, which play a role in maintaining the resting membrane potential of potassium ions close to equilibrium. KCNJ15 is known to be frequently expressed in the brain, kidney, lung, pancreas, and whole blood (Gosset et al., 1997). In humans, the synonymous SNP (rs3746876, C566T) located in the exon 4 of KCNJ15 is reported to be closely related to type II diabetes (Okamoto et al., 2010). In the present study, the three genes mentioned above were selected as the positional candidate genes of the QTL detected in SSC13. Based on this, the association between the SNPs located in their exonic region and the RBC, MCV, and MCH levels in pigs were examined in terms of statistical significance.
Genotype and statistic analysis
Next generation sequencing performed using the genomic DNA pooled from the two parental lines, and detected the SNPs that were located in the exonic region of the three genes and they exhibited differences between the two breeds. Ten SNPs were detected, all of which were confirmed as synonymous SNPs. To analyze their genotypes, eight pairs of specific primers were produced (Supplementary Table S2), and a genotype analysis was performed on the resource population of 1,232 pigs. Before an association test with three genetic traits, the frequencies of the genotypes were measured (Supplementary Table S3). In the KNP parental line, the genotypes of DYRK1A-SNP 1 (c.2989 G<A) and DYRK1A-SNP2 (c.4332 C<T) were fixed as “AA” and “TT,” respectively. For the Landrace parental line, the genotypes of DYRK1A-SNP3 (c.5070 C<A), KCNJ15-SNP1 (g.-293 C<G), KCNJ15-SNP2 (g.-207 G<A), and KCNJ15-SNP3 (g.1029 T<C) were fixed as “AA”, “GG”, “AA”, and “CC,” respectively. Their MAFs ranged from 0.0431 to 0.4397 (Supplementary Table S3).
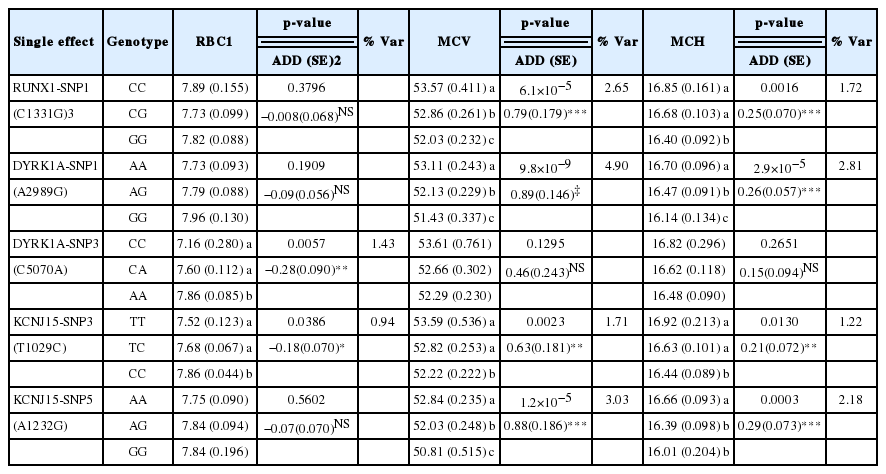
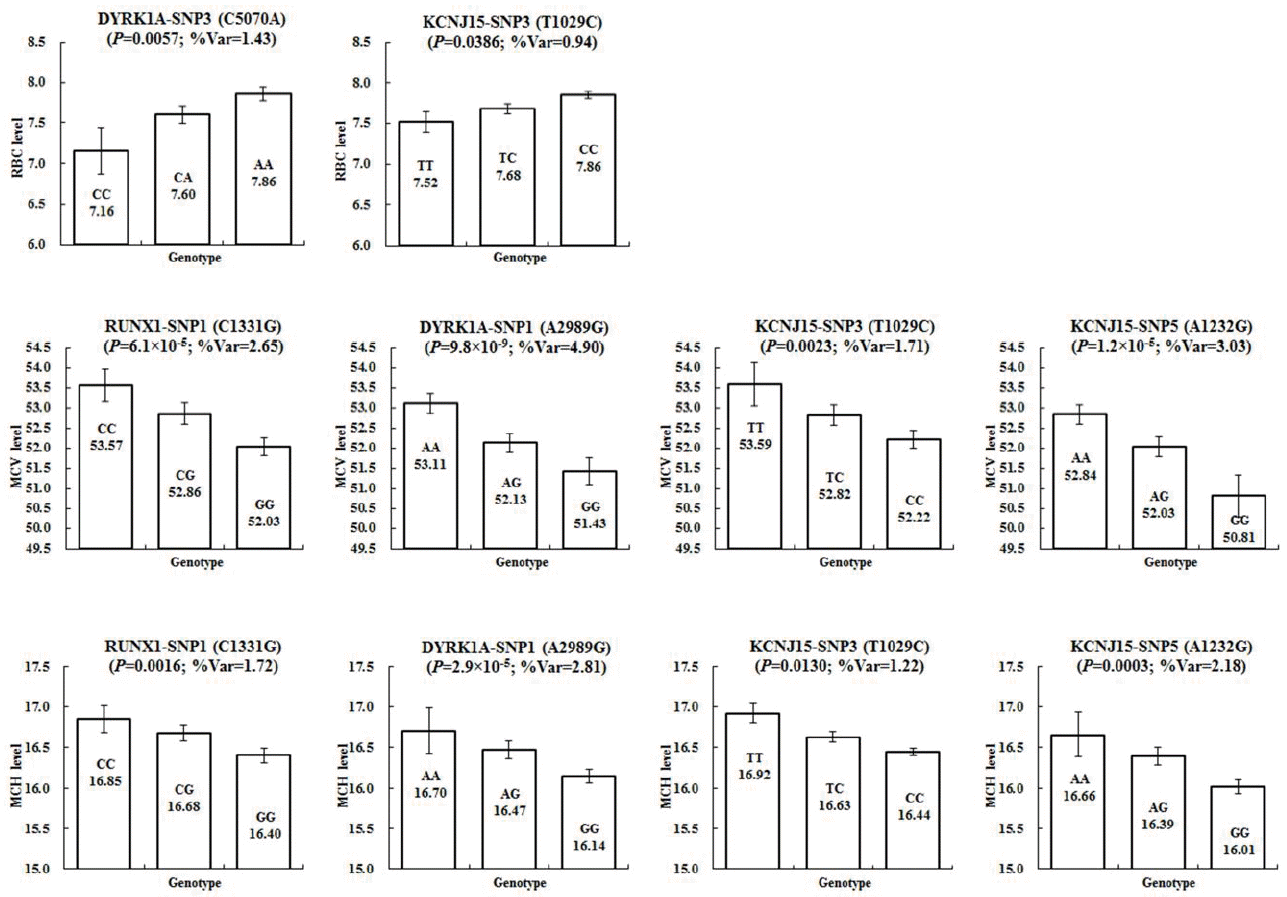
In this study, a verification test was performed on the association between the genotypes and the RBC, MCV, and MCH levels in pigs (Table 2). In the association analysis between the genotypes and the RBC level, DYRK1A-SNP3, and KCNJ15-SNP3 showed statistically significant results (Figure 1A). DYRK1A-SNP3 was found to have a nominal p value of 0.0057, which explained 1.43% of the phenotypic variance. Its additive effects exhibited negative values, indicating that the “A” allele inherited from Landrace is related to higher phenotypic values when compared to KNP. KCNJ15-SNP3 was explained as 0.94% of the phenotypic variance. Similarly to DYRK1A-SNP3, its additive effects showed an additive pattern of inheritance and negative values. For the remaining SNPs, other than the above two, their statistical significance in terms of the association between genotypes and genetic traits could not be detected (p>0.05).
Genotypic effects of the exonic variants SNPs in RUNX1, DYRK1A, and KCNJ15 on the pig RBC, MCV, and MCH levels in the pigs
The bar graph represents the genotypic mean and error of (A) red blood cell (RBC), (B) mean corpuscular volume (MCV), and (C) mean corpuscular hemoglobin (MCH) levels in the F2 generation of an intercross between Landrace and Korean native pigs. Significant associations (i.e., p value) between the genotype of single-nucleotide polymorphism (SNP) and traits, and additive genetic variances (% Var) explained by the SNP are given.
The analysis of the association between the genotypes and the MCV level showed that RUNX1-SNP1, DYRK1A-SNP1, KCNJ15-SNP3, and KCNJ15-SNP5 exhibited statistically significant correlations (Figure 1B). RUNX1-SNP1 showed a high level of statistical significance at a nominal p value of 6.1×10−5 and explained 2.65% of the phenotypic variance. The phenotype values corresponding to the three genotypes revealed statistically significant differences. With regard to the MCV level, DYRK1A-SNP1 was identified with the highest level of statistical significance (nominal p = 9.8×10−9) and explained 4.90% of the phenotypic variance. Moreover, it showed an additive pattern of inheritance. The “A” allele inherited from KNP through the additive effects of positive values was found to have higher MCV levels compared to the “G” allele inherited from Landrace. In the KCNJ15 genes, SNP3 and SNP5 were confirmed to have statistical significance and accounted for up to 3.03% of the phenotypic variance. All SNPs that were confirmed to be statistically significant in relation to the MCV level followed an additive pattern of inheritance and exhibited positive values. All the SNPs other than KCNJ15-SNP3 showed statistically significant differences in the phenotype values according to the three genotypes.
In the analysis of the association between the genotypes and the MCH level, similarly to the MCV level, RUNX1-SNP1, DYRK1A-SNP1, KCNJ15-SNP3, and KCNJ15-SNP5 were revealed to have statistical significance (Figure 1C). Among them, DYRK1A-SNP1 detected the highest level of statistical significance (nominal p = 2.9×10−5) and explained 2.81% of the phenotypic variance. RUNX1-SNP1, KCNJ15-SNP3, and KCNJ15-SNP5 accounted for up to 2.18% of the phenotypic variance. Similarly to the MCV level, their additive effects had positive values, and the allele inherited from KNP was related to higher phenotypic values. For DYRK1A-SNP3, which showed a statistically significant association with the RBC level, the statistical significance in relation to the MCV and MCH levels could not be detected.
In the body, blood performs gas exchanges, excretory functions, hormone delivery, and defensive activities. The measurement of the RBC, MCV, and MCH levels plays a key role in the diagnosis and classification of anemia (microcytic, normocytic, or macrocytic) (Bain, 1995; Freiberg, 2012). As this study did not measure the hematological values of the KNP and Landrace, their direct comparison was not possible. However, based on the results of a single marker association test on the positional candidate genes, KNP is predicted to have a lower RBC level and higher MCV and MCH levels than Landrace. This is in accordance with the results of our previous study, in which RBC, MCV, and MCH QTL followed additive patterns of inheritance, and the additive effect of QTL affecting RBC showed negative effects while QTL for MCV and MCH showed positive effects (Cho et al., 2011).
It is not possible to conclude that only those significant SNP in these results control the RBC, MCV, and MCH level in pigs. However, each genotype contributed to the levels of genetic traits in the F2 progeny in a resource population between Landrace and KNP. They will facilitate the disease-resistant breeding of pigs and serve important roles in providing a genetic basis for improving the robustness and health of pigs as well as humans. The present findings are also likely to provide follow-up studies with useful basic data.
Supplementary Information
ACKNOWLEDGMENTS
This work was carried out with the support of “Cooperative Research Program for Agriculture Science and Technology Development (Project No.PJ009971032016)” Rural Development Administration, Republic of Korea.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.