Characterization of the Fecal Microbial Communities of Duroc Pigs Using 16S rRNA Gene Pyrosequencing
Article information
Abstract
This study characterized the fecal bacterial community structure and inter-individual variation in 30-week-old Duroc pigs, which are known for their excellent meat quality. Pyrosequencing of the V1–V3 hypervariable regions of the 16S rRNA genes generated 108,254 valid reads and 508 operational taxonomic units at a 95% identity cut-off (genus level). Bacterial diversity and species richness as measured by the Shannon diversity index were significantly greater than those reported previously using denaturation gradient gel electrophoresis; thus, this study provides substantial information related to both known bacteria and the untapped portion of unclassified bacteria in the population. The bacterial composition of Duroc pig fecal samples was investigated at the phylum, class, family, and genus levels. Firmicutes and Bacteroidetes predominated at the phylum level, while Clostridia and Bacteroidia were most abundant at the class level. This study also detected prominent inter-individual variation starting at the family level. Among the core microbiome, which was observed at the genus level, Prevotella was consistently dominant, as well as a bacterial phylotype related to Oscillibacter valericigenes, a valerate producer. This study found high bacterial diversity and compositional variation among individuals of the same breed line, as well as high abundance of unclassified bacterial phylotypes that may have important functions in the growth performance of Duroc pigs.
INTRODUCTION
Animals harbor gut microbial communities whose composition and relative proportions of dominant microbial groups vary among species (Richards et al., 2005). The gut microbiota is believed to influence many metabolic processes, nutrient absorption, and the state of health of the host (Richards et al., 2005). Previous studies reported differences in the diversity and composition of the microbial community among swine breeds (Pajarillo et al., 2014a; Yang et al., 2014). In pigs, one of the most advantageous purebred lines commonly used for crossbreeding is the Duroc line, which shows superior growth performance (Schwab, 2007). Duroc is also known for excellent meat quality, tenderness, improved flavor, and palatability. These characteristics and performance attributes might be affected by metabolic processes in the gut along with the resident microbiota (Richards et al., 2005; Lamendella et al., 2011). Several reports have established the gut microbiota as a significant contributor to the digestion of food substrates that influences the overall physiological growth, immunologic responses, and pathogenesis in the host (Richards et al., 2005; Dowd et al., 2008).
However, profiles of the swine fecal microbiota have been limited to microorganisms that can proliferate in growth media under laboratory conditions. Culture-independent methods have identified various bacteria that are fastidious and recalcitrant to laboratory conditions (Richards et al., 2005). Recently, differences in the fecal microbiota among various pig breeds were investigated using polymerase chain reaction (PCR)-denaturing gradient gel electrophoresis (DGGE) profiling (Yang et al., 2014). However, the results were limited to organisms with high copy numbers, including Firmicutes, Bacteroidetes, methanogens, and sulfate-reducing bacteria; thus, the ability to characterize the overall microbial composition was limited. High-throughput pyrosequencing methods have enabled researchers to generate a massive amount of data for community analyses to describe the effect of weaning (Pajarillo et al., 2014b), age (Kim et al., 2011), diet (Lu et al., 2013), and antibiotics (Kim et al., 2012) on the swine gut microbiome. Here, we describe the fecal bacterial diversity among Duroc pigs using 16S rRNA gene pyrosequencing and suggest core members of the microbial community, as well as uncultivable members of the adult Duroc gut microbiota.
MATERIALS AND METHODS
Animals and sampling
All animals and protocols used in this experiment were approved by Dankook University Animal Care and Use Committee (Cheonan, Republic of Korea).
Seventeen vaginally delivered male Duroc pigs were used in this experiment. The pigs were weaned 4 weeks after birth and were given the same feed primarily based on corn and soybean meal (Supplementary Table 1). The daily feed allotment was offered as two meals at 12 h intervals (8:00 am and 8:00 pm). No antibiotics or feed additives were administered during the entire study. All animals were housed in an environmentally controlled room with a slatted plastic floor. Each pen was equipped with a 1-sided self-feeder and a nipple water-feeder to let the pig have access to feed and water ad libitum throughout the experimental period. The housing condition is at room temperature (25°C) and with 60% humidity, which also included a mechanical ventilation system and artificial light for 12 h using fluorescent lights. All rectal grab fecal samples were collected aseptically from individual pigs on the same day at 30 weeks of age. The fecal samples were immediately transported to the laboratory in a refrigerated container (4°C). After arriving to the laboratory, total genomic DNA was isolated from a 0.5-g aliquot of each fecal sample using a MoBio UltraClean Fecal DNA Isolation Kit (Carlsbad, CA, USA).
Pyrosequencing
Each DNA sample was used for PCR with barcoded primers flanking the V1–V3 hypervariable regions of 16S rRNA genes (http://www.ezbiocloud.net/oklbb/1001). Two units of ExTaq polymerase (Takara Bio Inc., Otsu, Japan) were used for PCR. The cycling parameters were as follows: initial incubation at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s and 55°C for 45 s, with a final extension at 72°C for 1 min 30 s. The products were separated by 1.5% (w/v) agarose gel electrophoresis and purified using Gel Extraction Kits (Macherey-Nagel, Düren, Germany) following the manufacturer’s instructions. Pyrosequencing was performed using Roche 454 GS-FLX Titanium chemistry (454 Life Sciences, Branford, CT, USA).
Data and statistical analysis
Raw sequence reads from each fecal sample were analyzed as described previously (Pajarillo et al., 2014a, b). Briefly, demultiplexing and removal of the barcodes were performed for all sequence reads. To minimize the effects of random sequencing errors, all trimmed sequences were subjected to quality control. We eliminated sequence reads that contained ambiguous base calls and those with fewer than 300 bases, and chimera filtration using the Bellerophon method, a partial-treeing approach, as well as the removal of unmatched sequences in the EzTaxon-e database (http://www.eztaxon-e.org) and a BLASTN search were performed.
Each valid pyrosequencing read was taxonomically assigned using the EzTaxon-e database (Chun et al., 2007). The following cut-off values were used for taxonomic assignment: species (x≥97%), genus (97%>x≥94%), family (94%>x≥90%), order (90%>x≥85%), class (85%>x≥80%), and phylum (80%>x≥75%), where x corresponds to the sequence identity between sequences within a certain operational taxonomic units (OTU) (Chun et al., 2007). The bacterial diversity of the microbial communities was calculated using sequence reads from individual samples (pigs) with OTUs defined up to 97% identity. The bacterial community composition and abundance were determined using CLCommunity software (ChunLab Inc., Seoul, South Korea). Percent abundances were calculated from the total valid reads for each pig fecal sample.
The following statistical analyses were performed in R version 3.1.0. Mean relative abundances and the standard deviation for the relative abundance of each taxon at the phylum, class, and genus levels were calculated and plotted as bar graphs and error bars, respectively. A heat map (heatmap[vegan]) showing the percent abundances of the top five taxonomic groups from the phylum level to the genus level was created for all samples. The variance caused by each taxonomic group was also calculated using the formula
where s2 is the sample variance, n is the sample size, x represents each individual measurement, and x\bar represents the sample mean (Politis and Romano, 1993). For phylogenetic tree reconstruction of an uncultured bacterium, the partial 16S rRNA gene sequence was aligned using ClustalW2 (Thompson et al., 1994) using the default parameters. Next, phylogenetic trees were constructed using the neighbor-joining method in MEGA5 (Tamura et al., 2011). The stability of the nodes was tested by bootstrap analysis using the adjusted values of 1,000 replicates.
RESULTS AND DISCUSSION
Although the Duroc line is an important pig breed with many favorable features (Schwab, 2007), data on its characteristics from an ecological perspective (e.g., gut microbiota) are limited. High-throughput 16S rRNA gene pyrosequencing offers a more qualitative and quantitative overview of microbial communities than other culture-dependent and -independent techniques, thereby providing substantial insight into the presence of both known and unclassified bacteria in the microflora. Previous studies have focused on the beneficial effects of the gut microbiota as it correlates with pig performance and growth (Richards et al., 2005). Multiple factors are believed to influence the overall health and performance of pigs, including the host genotype or breed type (Schwab, 2007), diet type (Hong et al., 2012), feed additives and antibiotics (Park et al., 2014), and farm management (Georgsson and Svendsen, 2002; Brossard et al., 2009).
Bacterial diversity in the Duroc line
The 17 samples generated a total of 108,254 valid sequence reads corresponding to 508 OTUs (95% identity) as indicated (Table 1 and Supplementary Tables 2 and 3). The Shannon indices ranged from 4.63 to 7.18, indicating notable diversity in terms of the evenness and richness of the species, which also showed inter-individual variation among bacterial communities as shown by varied diversity indices and richness estimates, though individuals are of the same age, breed, and diet. Nevertheless, the calculated diversity indices were significantly higher in this study than those in a previous report (Yang et al., 2014) using DGGE. These results indicate that pyrosequencing may provide more qualitative and quantitative information compared to other molecular-based techniques, including DGGE. It should be noted that high-level diversity and richness is beneficial to the host animal (Canibe et al., 2005) because it can contribute to various unique metabolic features or functionalities (Richards et al., 2005; Lamendella et al., 2011), leading to a more stable environment and reduced enteropathogen proliferation in the gut (Canibe et al., 2005).

Summary of the pyrosequencing data and diversity indices of 30-week old Duroc pigs (n = 17) at species level (OTU definition at >97% identity cutoff)
Microbial composition and the core microbiome
This study identified 29 phyla (Figure 1A), dominated primarily by Firmicutes (54.5%) and Bacteroidetes (39.7%) as expected for pigs of this age (Kim et al., 2011). Similar to our previous findings, these abundant phyla were shared with Yorkshire and Landrace (Pajarillo et al., 2014a). Other less abundant phyla detected in this study included Lentisphaerae (2.1%), Proteobacteria (1.4%), Tenericutes (1.4%), Spirochaetes (0.6%), and 23 other phyla comprising 0.2% of the total sequences analyzed. Clostridia (53.3%) dominated at the class level (Figure 1B), while Ruminococcaceae (34.5%) was dominant among the various families identified (Supplementary Table 4). At the genus level, Prevotella (12.8%) led the 89 taxa that were assigned to an identified genus (Figure 2A), while DQ906034_g (11.9%) was dominant among the 419 unclassified taxa (Figure 2B). The importance of Prevotella species is associated with their ability to degrade mucin (Mosenthin, 1998) and plant-based carbohydrates such as hemicellulose and xylans (Lamendella et al., 2011); this might be essential in feed digestion. These bacterial taxa, which have been consistently identified in pigs, should be studied from the physiological and morphological perspectives of the host.
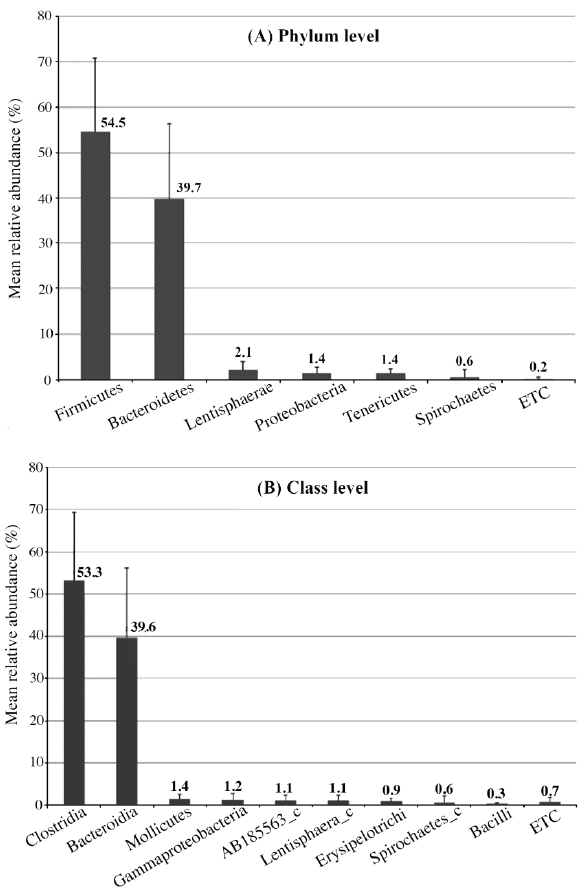
Bacterial composition and abundance of the fecal microbiota of Duroc (n = 17) pigs at the phylum (A) and class levels (B). The bar graph depicts the mean relative abundance of each taxonomic group in the pigs. Taxonomic groups with a calculated abundance <0.3% were pooled and labeled as ETC. The error bars represent standard deviations.
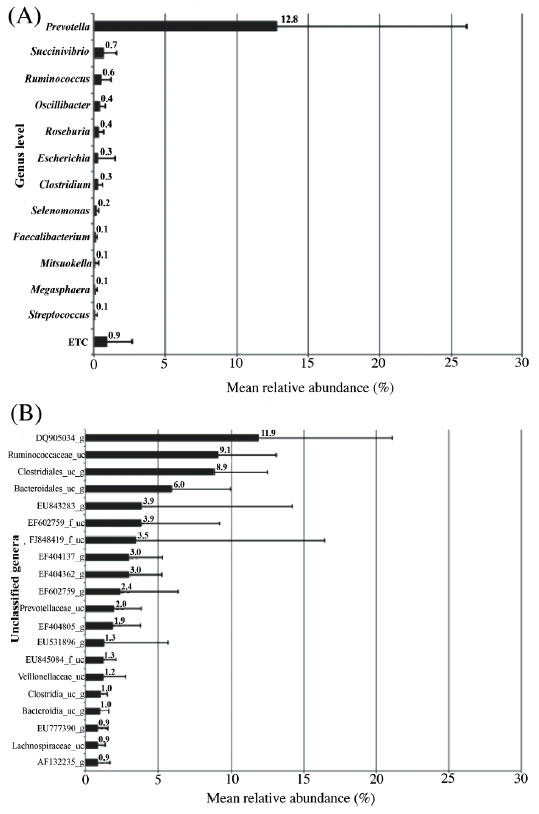
Mean relative abundance of the observed genera (A) and unclassified bacteria (B) in the pig fecal samples. Only bacterial phylotypes that had >0.1% of the total sequence reads are shown in the graph. Bacterial phylotypes that had <0.1% were pooled and labeled as ETC. The error bars represent standard deviations.
We also identified bacterial OTUs (95% identity) corresponding to core bacteria that may play a key role in maintaining the metabolism of the gut in Duroc pigs (Kim et al., 2011; Pajarillo et al., 2014a, b). Bacterial genera, including Prevotella, Succinivibrio, Ruminococcus, Roseburia, and Clostridium, as well as unclassified OTUs (95% identity), including DQ905034_g, EU843283_g, EF404137_g, EF404362_g, and EF602759_g (Supplementary Tables 2 and 3), were identified as the possible core fecal microbiome in Duroc because these bacteria were present in all adult Duroc pig fecal samples (n = 17) regardless of their abundance in each individual. Among the core bacterial genera, Prevotella has been reported to dominate the swine gut (Lamendalla et al., 2011; Lu et al., 2013), particularly in the Duroc breed (Pajarillo et al., 2014a). Succinivibrio, on the other hand, was previously detected in weaned and growing pigs (Kim et al., 2011; Pajarillo et al., 2014b). Roseburia species were seen in Lactobacillus plantarum-treated pigs using DGGE (Pieper et al., 2009), weaned pigs (Pajarillo et al., 2014b), and growing pigs (Kim et al., 2011). Clostridium frequents the ileum of pigs (Dowd et al., 2008) and was found to be associated with Lactobacillus plantarum-treated pigs using DGGE (Pieper et al., 2009). Likewise, Ruminococcus is among the genera reported to be a frequent inhabitant of the ileum in pigs (Dowd et al., 2008). These core bacteria of the Duroc breed should be studied in association with the host’s genome, transcriptome, and metabolome, as well as their function in the growth and performance of Duroc pigs (i.e., daily weight gain, feed efficiency, and response to antibiotic growth promoters).
Among the unclassified OTUs having 95% identity (Figure 2B and Supplementary Table 3), DQ905034_g was found to be present in all pig fecal samples and had the highest recorded abundance among several unclassified OTUs detected in Duroc feces. A phylogenetic analysis of the representative sequence revealed that it is closely related to Oscillibacter valericigenes strain Sjm18–20 (Supplementary Figure 1), a representative bacterium in the Oscillibacter group capable of producing valerate (Iino et al., 2007), which is an important short-chain fatty acid associated with certain health benefits due to its ability to replace butyrate as the energy source for colonocytes in D-tagatose fermentation in pigs (Lærke and Jensen, 1999) and higher feed efficiency in cattle (Guan et al., 2008), as well as its ability to inhibit cell proliferation and increase alkaline phosphatase activity in human adenocarcinoma cell lines (Siavoshian et al., 1997). The high abundance of this bacterium suggests its importance to pig health, particularly gut functionality and metabolic activity. The ability to increase certain metabolites through supplementation with specific bacteria may be possible without changing the composition of feed. However, further investigation of the association of this bacterium with the growth performance or meat quality of the Duroc line is required.
Inter-individual variation in swine microbiota
As described above, variation in bacterial diversity was found even among individual pigs of the same purebred line, diet, and age. In terms of bacterial composition, we observed inter-individual variation, starting at the family level (Figure 3), which was supported by data produced at each taxonomic level (Supplementary Table 4). High variance indicates differences among individual samples, while a low variance points to homogeneity. Though the microbial community structure associated with the host may appear to be uniform among samples, inter-individual variation in the microbiome is still apparent and is said to be specific and functionally relevant (Richards et al., 2005; Lamendella et al., 2011).

Heat map showing the relative abundance of the five most abundant taxonomic groups in the pigs. Depicted in the plot are individual pigs (n = 17) containing the most abundant groups from the phylum to the genus level. The relative levels of abundance are depicted visually from black to white; black represents the lowest abundance (min = 0%), while white (max = 100%) represents the highest level of abundance.
Previous studies showed inter-individual variation in human and animal microbiomes. The intestinal microbiota of neonates and premature infants (Mshvildadze et al., 2010), vaginal microbiota (Zhou et al., 2009), and skin microbiota (Gao et al., 2007) have high inter-individual diversity and fluidity as key characteristics of their microbial populations. An analysis of the gut microbiota in a probiotic-fed fish population and the fecal microbiota of horses showed that bacterial populations are influenced by day-to-day fluctuations and inter-individual differences (Ige, 2013; Dougal et al., 2014). Interestingly, though inter-individual variation has been observed in human and animal microbiome studies, the metabolic functions of the microbial community associated with the host are still believed to be conserved (Gao et al., 2007; Zhou et al., 2009; Lamendella et al., 2011). It is thus important to investigate what factor drives inter-individual variation in a controlled environment as well as the extent of its effect on and association with animals, particularly with pigs of the same purebred line.
Many feeding trials have shown significant variation in animal responses or growth performance between individuals of the same treatment group (Georgsson and Svendsen, 2002; Brossard et al., 2009). This indicates that the intestinal microflora of each individual animal may react differently to the same feed. The complexity of the gastrointestinal tract might drive these variations between animals, exerting different effects on individual animals (Ige, 2013). This should be an important consideration during feed development and feed additive formulation in the future.
CONCLUSION
This study described the diversity and composition as well as inter-individual variation in the overall fecal bacterial populations of Duroc pigs raised in environmentally controlled facility. High-throughput 16S pyrosequencing of the V1–V3 hypervariable regions of the 16S rRNA successfully described and provided a substantial insight on the presence of both known bacteria and unclassified fecal bacterial flora. Among the core microbiome observed, Prevotella genus which is a significant contributor in mucin degradation and carbohydrate digestion, and an unclassified OTU Oscillibacter valericigenes (DQ905034_g) predominated in the feces. Further functional characterization of these bacteria is essential for developing animal probiotics in the future.
Supplementary Data
ACKNOWLEDGMENTS
This work was supported by a grant from the Next-Generation BioGreen 21 Program (PJ00812701), Rural Development Administration, Republic of Korea. The present research was also conducted by the research fund of Dankook University in 2013.