Sex Determination of Cattle Meat by Polymerase Chain Reaction Amplification of the DEAD Box Protein (DDX3X/DDX3Y) Gene
Article information
Abstract
Determination of sex origin of cattle meat by fast and reliable molecular methods is an important measure to ensure correct allocation of export refunds particularly in European countries and also female cattle (cow) slaughter is legally banned in India because of religious beliefs. Based on the DEAD box protein gene located on the X and Y chromosomes, 2 pair of primers were designed and the system of PCR was optimized. Upon PCR amplification, male tissue showed 2 bands, while female tissue resulted in only one band. The accuracy and specificity of the primers was assessed using DNA template extracted from cattle meat of known sex. The protocol was subjected to a blind test and showed 100% concordance, proving its accuracy and reliability.
INTRODUCTION
Determination of sex origin of cattle meat has been always of public interest in country like India where slaughter of cow (female cattle) is banned because of religious beliefs and laws thereby. The economic aspect allied with such discriminatory slaughter policy also gets support as male beef is designated to be of higher quality than cow meat and therefore yield higher prices (Zeleny and Schimmel, 2002) particularly in European countries. To implement the regulations pertaining to such issues and to assure consumers of accurate labeling meat analysts need to have reliable, authentic and simple method for determining the sex of cattle meat.
To date, a range of different methodologies have been developed for determining the sex of meat, mainly based on detecting either hormone or DNA (Zeleny and Schimmel, 2002). In general terms, hormone-based methods include immunochemical determination using ELISA (Simontacchi et al., 1999), and chromatographic techniques with mass-spectrometric detection, such as high performance liquid chromatography-mass spectrometry/mass spectrometry (HPLC-MS/MS) (Draisci et al., 2000) and gas chromatography-mass spectrometry (GC-MS) (Hartwig et al., 1997). Most of the methods proved to be highly specific and sensitive, but were not performed on a regular basis for meat sexing due to the technical limitations or the expensive equipments required.
Over the last few decades, DNA-based techniques, especially polymerase chain reaction (PCR)-based methods for meat sexing have received particular attention, and have proved to be reliable, sensitive, and fast (Tagliavini et al., 1993; Appa Rao et al., 1995). DNA regions that differ between male and female individuals are essential features in PCR sex determination. In general, these methodologies were developed mainly based on amelogenin gene (AMELX and AMELY) (Ennis and Gallagher, 1994; Chen et al., 1999; Fontanesi et al., 2008), zinc finger gene (ZFX and ZFY) (Aasen and Medrano, 1990; Kirkpatrick and Monson, 1993; Zinovieva et al., 1995; Lockley et al., 1997) and the sex determination region of the Y chromosomal gene (SRY) (Fu et al., 2007; Lu et al., 2007; Shi et al., 2008) Alternatively, the bovine-specific repetitive sequence BRY-1 (Matthews, 1990) and the single copy sequence BOV97 M (Miller and Koopman, 1990) are also used in the analysis for the sexing of cattle. More recently, based on real time PCR both SYBR Green (Ballin and Madsen, 2007) and TaqMan (Parati et al., 2006) for bovine sex determination have also been reported.
The advantage with AMEL gene, zinc finger gene and DEAD box protein gene over Y chromosomal gene (SRY) is their presence on both sex chromosomes, because there is need of developing the suitable strategy for differentiating the negative amplification from no amplification due to PCR failure especially when Y chromosome specific primers are to be used. However the amplified product size and the difference between male and female in case of AMEL gene are larger whereas in case of zinc finger gene, differentiation of male and female sex requires RFLP technique. The DEAD box protein gene seems to be unique alternative because of easy amplification with a moderate difference in amplified product size between male and female. The purpose of the present work was to develop a protocol for sexing of cattle meat based on the DEAD box protein gene (DDX3X/DDX3Y) using PCR technique.
MATERIALS AND METHODS
Sample selection and DNA extraction
A total of 16 muscle tissue samples of cattle (8 male and 8 female) were collected from Referral polyclinic, Indian Veterinary Research Institute, Izatnagar. Further, a total of 12 meat samples (6 male and 6 female) were collected from local market of Kerala and West Bengal, India. The collected samples were transported to the laboratory under refrigeration, and were stored frozen at −20°C prior to analysis. Genomic DNA was extracted from the samples using the DNeasy® blood and tissue kit (QIAGEN, Germany) according to the manufacturer’s instructions. Subsequently, the quality of genomic DNA was assessed by agarose gel electrophoresis using 0.8% agarose gel (AMRESCO, USA) stained with ethidium bromide. The purity and concentration of DNA was estimated spectrophotometrically using Nanodrop® ND-1000 spectrophotometer (Thermo Scientific) at 260 and 280 nm. The DNA sample showing the OD260:280 nm value of 1.70–1.90 was considered as good quality.
Design of PCR primers
As target for PCR primers the DEAD box protein gene was chosen in this study. Based on the information obtained from the alignment of NCBI Reference sequences of the DDX3X gene (GenBank Accession No. NW_003104743.1) and DDX3Y gene (GenBank Accession No. NW_003104786.1) sequences, two pairs of primers DDX3-1F (AGGAAGCCAGGAAAGTAA), DDX3-1R (CATCCA CGTTCTAAGTCTC) and DDX3-2F (TGAGGAAGCCA GGAAAGTAAGTAT), DDX3-2R (GCACCACCRTAWA CCACACAA) were designed with homology between X and Y chromosome. DDX3-1F and DDX3-1R primers were expected to yield a PCR fragment of 208-bp with the DDX3X gene as target sequence and 184-bp with DDX3Y gene, respectively. DDX3-2F and DDX3-2R primers were expected to yield a PCR fragment of 171-bp with the DDX3X gene as target sequence and 147-bp with DDX3Y gene, respectively (Figures 1 and 2). The PCR primers were designed using GeneTool® software and checked the hairpin structure and self ligation of these primers. The PCR primers were synthesized by Eurofins Genomics India Pvt Ltd, Bangalore.
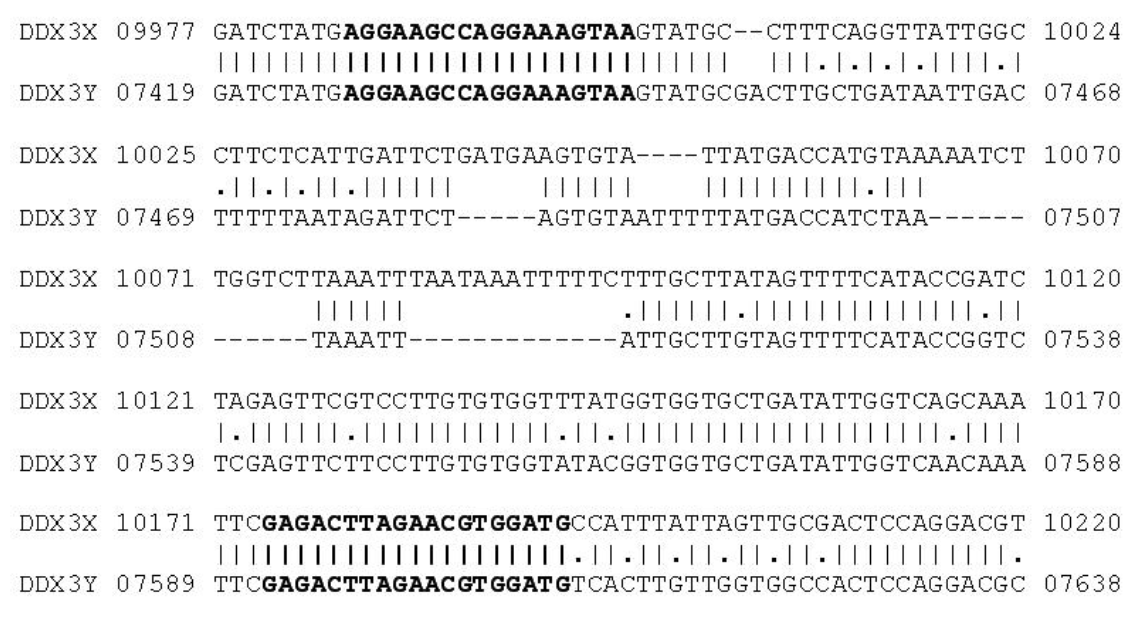
Alignment of the DDX3X and DDX3Y gene sequences (GenBank accession No. NW003104743.1 and NW003104786.1). The primers DDX3-1F and DDX3-1R are indicated by bold letters. Dashes indicate gaps in alignment.

Alignment of the DDX3X and DDX3Y gene sequences (GenBank accession No. NW003104743.1 and NW003104786.1). The primers DDX3-2F and DDX3-2R are indicated by bold letters. Dashes indicate gaps in alignment.
PCR amplification
Polymerase chain reaction (PCR) was performed in 25 μl of reaction mixture containing 50 ng of genomic DNA, 200 μm of each dNTP, 1.5 mM MgCl2, 5 pmoles of each DDX3-1F and DDX3-1R primers, 1 unit GoTaq® Flexi DNA Polymerase (Promega, Madison, WI, USA), 1× PCR colored buffer (Promega, Madison, WI, USA) and nuclease-free water to make a final volume. Amplification was performed on a PTC-200 DNA Engine® thermal cycler, (Bio-Rad, USA) using 0.2 ml reaction tubes. The PCR programme consisted of 5 min denaturation at 94°C, followed by 34 cycles of denaturation (94°C, 45 s), annealing (53°C, 45 s) and primer extension (72°C, 60 s). The final cycle was followed by extension at 72°C for 10 min and indefinite hold time at 4°C. PCR conditions for DDX3-2F and DDX3-2R primers were also same as indicated above except annealing temperature (56°C).
Gel electrophoresis
The PCR product obtained was analyzed by agarose gel electrophoresis in 2% agarose gel (AMRESCO, USA) stained with ethidium bromide. A 100-bp DNA ladder (O’Gene Ruler, Fermentas) was electrophoresed simultaneously in order to assess the size of amplification product. The gel was visualized in automatic gel documentation system (MiniBis, DNR Bio-Imaging Systems) and the size of the amplicon was determined using software available with the gel documentation system.
RESULTS AND DISCUSSION
PCR products of cattle meat samples after electrophoresis showed two bands for tissue from male (208,184-bp bands for DDX3-1F/DDX3-1R primers and 171,147-bp bands for DDX3-2F/DDX3-2R primers), while female tissue resulted in only one band (208-bp band for DDX3-1F/DDX3-1R and 171-bp band for DDX3-2F/DDX3-2R primers) (Figures 3 and 4, respectively).

Electrophoretic pattern of amplified fragments from the cattle meat DNA using DDX3-1F and DDX3-1R primers. Samples are: male (Lanes 1, 3 and 5) and female (Lanes 2, 4 and 6). M-Marker of molecular weight, 100-bp ladder (O’Gene Ruler, Fermentas), N = Non-template control.
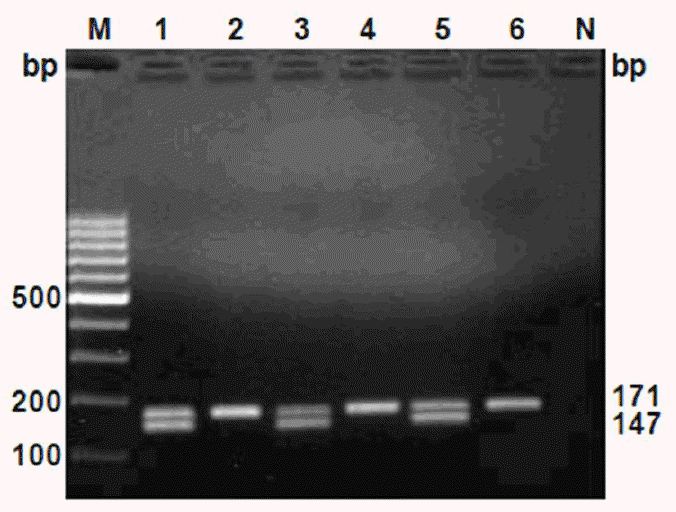
Electrophoretic pattern of amplified fragments from the cattle meat DNA using DDX3-2F and DDX3-2R primers. Samples are: male (Lanes 1, 3 and 5) and female (Lanes 2, 4 and 6). M-Marker of molecular weight, 100-bp ladder (O’Gene Ruler, Fermentas), N = Non-template control.
During the last few decades, based on PCR technique, several different methodologies were developed for determining the sex of domestic animal meat (Tagliavini et al., 1993; Appa Rao et al., 1995; Ballin and Madsen, 2007), most of which were thought to be reliable, sensitive, and fast (Zeleny et al., 2002; Zeleny and Schimmel, 2002). Male beef is designated to be of higher quality than cow meat because cow meat is usually derived from females, usually at the end of their useful reproductive life, or at the end of their time as milk producers (Tagliavini et al., 1993). Sexing of cattle meat is of potential value in the quality control, as well as, in protecting the interests of consumers. In this work, we have proposed a reliable, fast, and straight forward method for the sexing of cattle meat through PCR amplification of DEAD box protein gene using only one primer set with amplified product size of less than 200-bp and moderate difference of 24-bp between male and female.
DEAD box polypeptide 3 (DDX3) is a subfamily of the DEAD (for the sequence Asp-Glu-Ala-Asp) box RNA helicases, a family of proteins characterized by the conserved motif (Abdelhaleem, 2005; Rosner and Rinkevich, 2007). The DEAD box proteins are ATP-dependent, and involved in unwinding double-stranded RNA structures and remodeling RNA protein interactions. Thus, they are crucial for RNA metabolism, including transcription, splicing, RNA export, ribosome biogenesis, RNA degradation and translation initiation (Benz et al., 1999). In cattle, this gene is located on the X-chromosome (DDX3X) and the Y-chromosome (DDX3Y) (Liu et al., 2009). Due to length polymorphism between DDX3X and DDX3Y this gene is used as a target for sex determination in this study. Due to an insertion or deletion in the region amplified in the X-specific (DDX3X) or Y-specific (DDX3Y) gene, the amplified fragments are of different sizes.
The efficiency of these primers were tested on the male as well as female cattle meat samples, collected from two different states of India (i.e.) Kerala, West Bengal, which are approximately 2,000 km apart. The primers were effectively differentiated the sexes of the randomly coded samples for each set of primers (Figures 5 and 6), which suggests the wider applicability of these primers in sex determination of cattle meat.
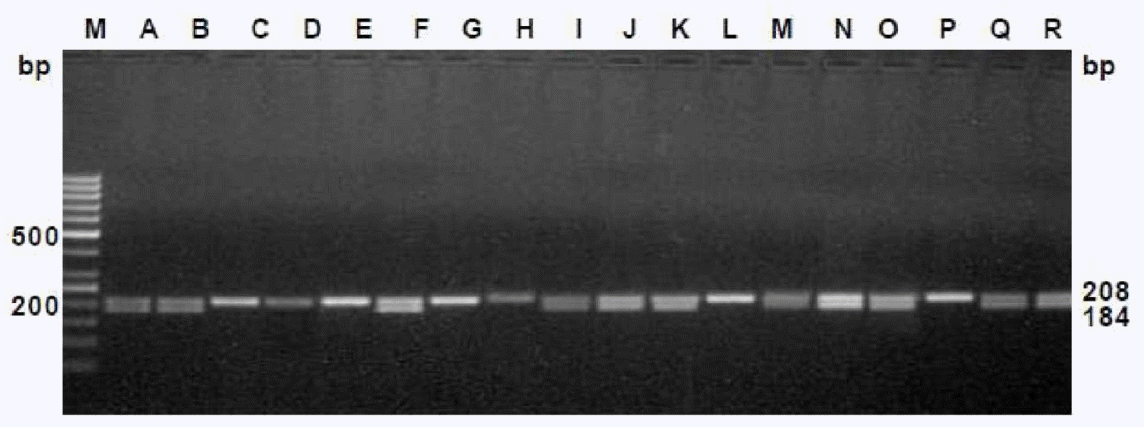
Laboratory validation of the assay in randomly coded samples using DDX3-1F and DDX3-1R primers. Samples are: male (Lanes A, B, F, I, J, K, M, N, O, Q and R) and female (Lanes C, D, E, G, H, L and P). M-Marker of molecular weight, 50-bp ladder (O’Gene Ruler, Fermentas).
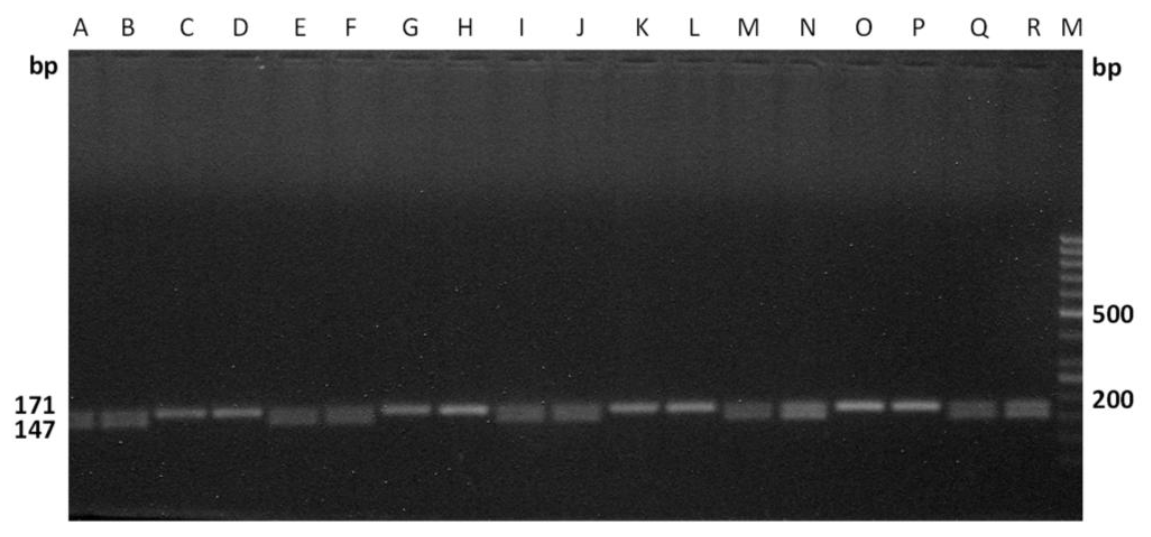
Laboratory validation of the assay in randomly coded samples using DDX3-2F and DDX3-2R primers. Samples are: male (Lanes A, B, E, F, I, J, M, N, Q and R) and female (Lanes C, D, G, H, K, L, O and P). M-Marker of molecular weight, 50-bp ladder (O’Gene Ruler, Fermentas).
CONCLUSIONS
In conclusion our findings show that the PCR assay based on the DEAD box protein gene is reliable for sex identification in cattle meat. Due to relatively short size (<200-bp) of the PCR products, this method can be successfully applied to sex determination of cattle meat samples with highly degraded DNA in a relatively shorter period of time. Also, the advantage of this assay is that neither additional control amplicons with a second locus-specific autosomal primer pair nor restriction endonuclease steps are necessary for sex determination and control of the PCR reaction. To our knowledge, this is the first report to demonstrate the applicability of DEAD box protein gene for sexing cattle meat.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Indian Veterinary Research Institute (IVRI) and Central Avian Research Institute (CARI), Izatnagar, India for providing necessary facilities to accomplish this research.