VCS: Tool for Visualizing Copy Number Variation and Single Nucleotide Polymorphism
Article information
Abstract
Copy number variation (CNV) or single nucleotide phlyorphism (SNP) is useful genetic resource to aid in understanding complex phenotypes or deseases susceptibility. Although thousands of CNVs and SNPs are currently avaliable in the public databases, they are somewhat difficult to use for analyses without visualization tools. We developed a web-based tool called the VCS (visualization of CNV or SNP) to visualize the CNV or SNP detected. The VCS tool can assist to easily interpret a biological meaning from the numerical value of CNV and SNP. The VCS provides six visualization tools: i) the enrichment of genome contents in CNV; ii) the physical distribution of CNV or SNP on chromosomes; iii) the distribution of log2 ratio of CNVs with criteria of interested; iv) the number of CNV or SNP per binning unit; v) the distribution of homozygosity of SNP genotype; and vi) cytomap of genes within CNV or SNP region.
INTRODUCTION
In genomic research, copy number variation (CNV) and single nucleotide polymorphism (SNP) are used to identify the association with complex phenotypes or susceptibility or resistance to diseases (O’Donovan et al., 2008; Glessner et al., 2009; Xu et al., 2009). The CNV encompasses more DNA than SNP and contains entire genes and their regulatory region (Feuk et al., 2006). The type of genetic variant can influence gene dosage other than phenotypic variation, which might cause genetic diseases. A series of studies using CNV and SNP were performed to detect the association with different cancers or diseases (Diskin et al., 2009; Shlien and Malkin, 2009). Development of whole genome sequencing projects of different organisms and the concurrent improvement in biotechnologies have contributed to the detection of enormous numbers of SNP and CNV in each species. Thousands of CNV and SNP are currently available in the public databases but it is not so easy for local researchers to use them for their own analyses. Information regarding CNV and SNP in general consists of numerical values which are difficult to understand and to interpret biologically. Visualization of the data may assist researchers to interpret biological meanings from the numerical value, even though it is not a necessary step for the analyses. However, few visualizing software have been reported for CNV and SNP. In this study, we developed a web-based visualization tool graphically representing the enrichment of genome contents in CNV, the distribution of CNV and SNP on chromosomes, the log2 ratio of fluorescence intensities of CNV, the homozygosity of SNP on chromosomes, and cytomapping of the genes of interest.
PROGRAM OVERVIEW
We developed a web-based tool called the VCS (visualization of CNV or SNP) for visualizing data of CNV or SNP in the genome, which consists of six main menus. The pictures can help not only to interpret a biological meaning from the numerical value of CNV or SNP but also provide the figures for user’s manuscript. The VCS tool provides a graphical view of the physical distribution of CNV or SNP on chromosomes. Although several web databases have reported annotated CNV (e.g. Database of Genomic Variants [DGV; http://projects.tcag.ca/variation/], dbSNP 131 [http://www.ncbi.nlm.nih.gov/], GWAS CENTRAL(http://www.gwascentral.org/), and SNP and CNV Annotation Database [SCAN; http://www.scandb.org/]) or CNV extraction software (e.g. PennCNV [Wang et al., 2007], Aroma.affymetrix [Bengtsson et al., 2008], CRLMM [Scharpf et al., 2010], and Affymetrix Power Tools [Lockstone, 2011]), it is often difficult to apply them one’s own result. Main features of VCS are as follows.
Enrichment genome contents visualization
The VCS shows the enrichment genome contents (gene, long interspersed nuclear element [LINE], short interspersed nuclear element [SINE], long terminal repeat [LTR], simple repeat, low complexity, miRNA, tRNA, CpG island, and Gene Ontology – Biological Process, Molecular Function, and Cellular Component) in region having specific range such as CNV. For cluster analysis, the distance matrix was produced by Hamming distance computation considering deletion and duplication of copy number. Then the hierarchical cluster and principal component analysis (PCA) were performed using the distance matrix. As the result, user can easily show the nearest clustered samples about the genome content within CNV region. The input file needs matrix format file formed 0, 1, 2, 3, 4. Here, 0 and 1 is deletion and more than 2 is duplication. The figure represents as user-defined such as deletion or insertion. So the user can show the enrichments result figure and table of genome content in a specific region (Figure 1A), and show hierarchical and PCA cluster among samples. In addition, user can display all the genome contents per sample. If user denotes groups as _A, _B, _C in input file, user can easily and clearly show clusters as editing the grouping image using other graphic tool such as Adobe photoshop or illustrate.
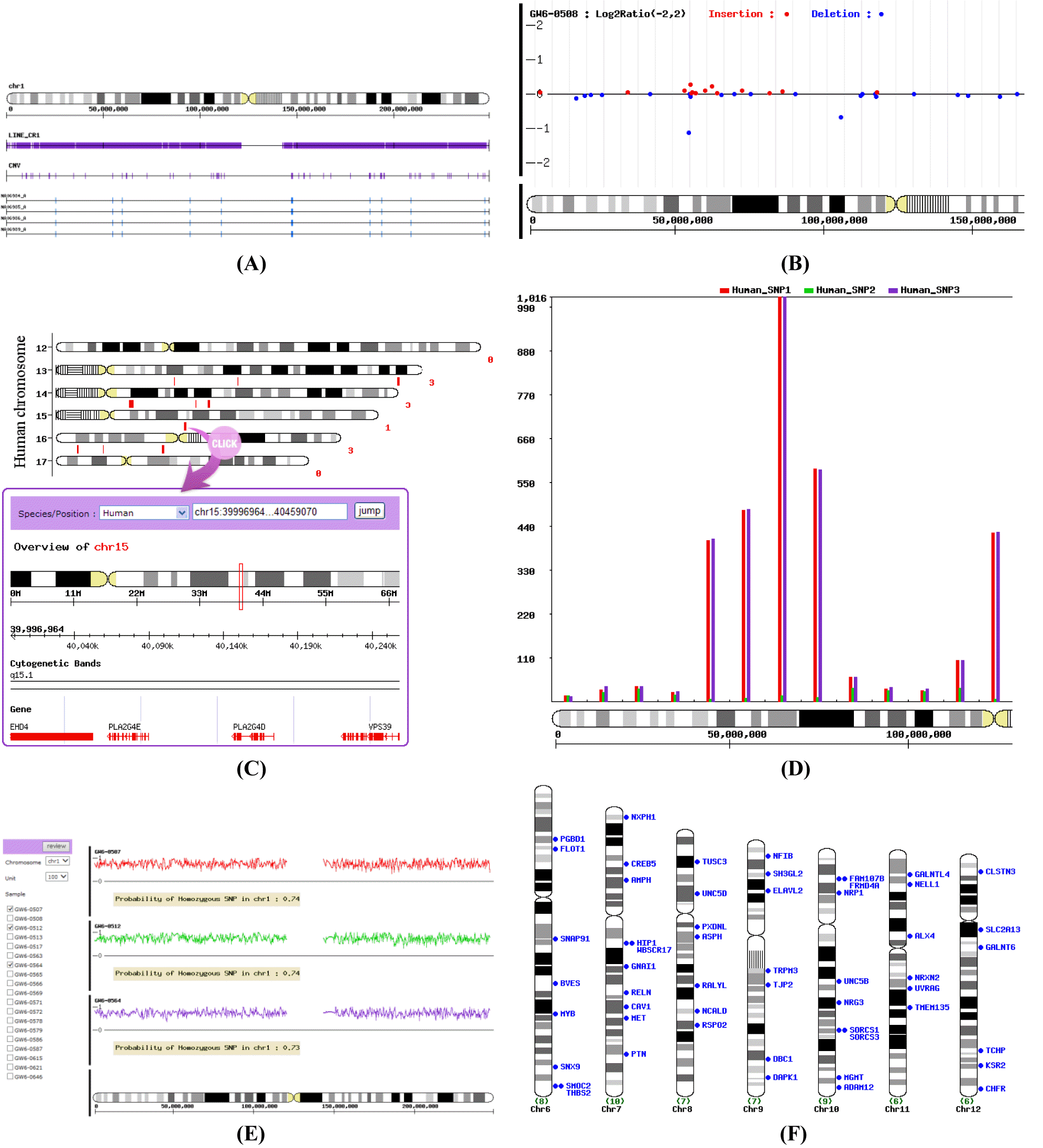
(A) Visualization of the enrichment of genome contents in CNV region, (B) visualization of the distribution of log2 ratio, with red (insertion) and blue (deletion) marks, (C) visualization of the physical distribution for specific position or region, (D) visualization of the distribution of SNP numbers per binning unit 1 Mb on chromosome, (E) visualization of the distribution of homozygous SNP. Irregular zig-zagged lines represent the homozygosity value per unit of 100 SNPs, (F) displays the CytoMap for genes located in the CNV or SNP subregion. CNV, Copy number variation; SNP, single nucleotide phlyorphism.
Physical distribution visualization
The VCS tool provides a graphical view of the physical distribution of CNV or SNP on chromosomes. Any marker contained information of chromosomal position by point (SNP) or specific ranges (CNV, miRNA, and repeat sequence) can be used in this tool (Kim et al., 2010; 2011; 2013). This menu is useful for comparing the physical distribution of your own CNV or SNP. In addition, comparison among samples is available by adding input files up to five. The input file simply needs the information of chromosome number and chromosomal position of either CNV or SNP. After your data are loaded on the website, you can obtain the information in detail on the genome where the CNV (SNP) is located by clicking it (Figure 1C). User can take a look at the information on genes, and repeat sequences such as SINE, LINE, LTR, and simple repeat around the CNV.
Log2 ratio distribution visualization
The VCS plots log2 ratio of CNV with insertions and deletions that are more conspicuous. The log2 values are plotted at the middle position of CNV regions across the chromosome. Several web databases represent the whole log2 ratio (e.g. Affymetrix Genotyping Console Browser), but VCS can provide the criteria which is the user-adjustable log2 ratio. So a user can create the view of CNV filtrated by adjusting the criteria with different log2 ratio values for different research purposes. In addition, user can draw a Manhattan plot which easily can define appropriate significance value, and can perform the comparison among samples selected in this menu. The input file needs matrix format data with the information of physical location and the value of plus (+) or minus (−) such as log2 ratio after CNV analysis. The VCS then gives the following output as user-defined criteria, from which you can obtain total counts and median size of gain (insertion), loss (deletion), and complex (insertion and deletion) (Figure 1B). Default of a criteria set up ±0.3 which is widely used in biology research. And you can show distribution of visualized log2 ratio and/or ±values.
Variation distribution visualization per binning unit
The VCS calculates the number of CNV or SNP per binning units of 10 kb, 100 kb, 1 Mb, and 10 M. The goal of this menu is to look at the number of variants within the certain ranges of physical distances, which allows researchers to take advantage of deciding or selecting the scale of the study area they want to focus on. Also, this menu is useful for comparing the numbers per binning unit by adding more data. The user selects binning unit by simply clicking on the appropriate criteria. The input file is the same input file with the information of chromosome number and chromosomal position of either CNV or SNP used for the physical location. You can show the visualized distribution per binning unit and decide concentrated study region on genome (Figure 1D).
Homozygosity distribution visualization for single nucleotide phlyorphism genotypes
The VCS shows the homozygosity of SNP on chromosomes by using the information of SNP genotypes of samples, chromosomal position of SNP and chromosome number. This menu is useful when comparing homozygosity among samples. The VCS calculates homozygosity of all SNP located on an entire chromosome of interest and plots homozygosity of every unit of 100 SNPs along the chromosomes. At the end of the chromosome, the number of SNP is usually less than 100 which is added to the previous unit if the number of SNP is ≤50 or is calculated as another unit if it is >50. The input file requires the matrix data with information such as genotypes in SNP analysis. User can then display any area that has a low homozygosity value, and obtain the probability of the homozygous SNP on each chromosome (Figure 1E).
CytoMap
CytoMap provides the cytomapping figure of your focused-genes (Figure 1F). The input file needs only the information of the cytoband of your focused-genes. There are several assembly versions of human genome sequences available in public databases such as National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) and University of California Santa Cruz (http://genome.ucsc.edu/). However, the physical positions of genes of interest are version-dependant. CytoMap provides a gene map by the cytoband position. This menu is useful for genome-wide view of data.
IMPLEMENTATION
The VCS application is available from http://snugenome.snu.ac.kr/Software/VCS/. Executable examples can be downloaded from the same web site as well. The VCS was implemented as a program written in PHP (PHP Hypertext Preprocessor) (ver.5.3), mysql (ver. 5.1.36) and Python (ver.2.5). Animal species included in this study are human (hg19), rhesus (rheMac2), mouse (mm9), rat (rn4), dog (canFam2), horse (equCab2), cow (bosTau4), opossum (monDom5), chicken (galGal3), zebrafish (danRer7), D. melanogaster (dm3), and C. elegans (ce6). Genomic information of those species was downloaded from http://genome.ucsc.edu/. By selecting a species from the pop-up menu, basic genomic information of the species such as total number of chromosomes and sizes of chromosome is set as a default for the analysis. Therefore, a user doesn’t need to prepare the information in the input file regardless of any platform such as Affymetrix or Illumina for analysis of either CNV or SNP. The input file only needs the information of chromosomal position or CNV log2 ratio values or SNP genotypes or cytoband after variation analysis. For each menu, input file format take the divided by tabs or comma. For output file, you can select formats: png or bmp. Also user can edit the image using other graphic tool such as photoshop or illustrate. A researcher who is interested in CNV or SNP can easily access the web site and use it for free without additional steps of downloading and installing it onto their local computer. This tool is user friendly and can be simply used without a thick user’s manual. To development of bioinformatics usages of the data served in VCS, we are continuously developing and updating. We expect to add tool associated with these CNVs and SNPs studies are merged into VCS.
ACKNOWLEDGMENTS
This work was supported by a grant from the Next-Generation BioGreen 21 Program (No. PJ008116), Rural Development Administration, Republic of Korea. All authors read and approved the final manuscript.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.