





INTRODUCTION
The domestic pig, Sus scrofa domestica, has long been an important source of dietary protein for humans and has undergone natural selection in response to various environmental factors over time. Furthermore, pig breeds have been subject to intensive artificial selection to increase economically important traits such as reproduction, growth rate, stress resistance, and meat quality [1,2]. In particular, Berkshire pigs have been under intensive artificial selection, particularly to ensure rapid and efficient muscle accumulation and desirable pork qualities [3]. As a result of strong selection, the quality of Berkshire meat is considered superior, as it contains a high proportion of neutral lipid fatty acids and good marbling fat [4], both of which are important factors contributing to palatability traits such as tenderness and juiciness [5]. These unique meat qualities, as well as biochemical and physical features, have been investigated in a sensory panel analysis [6ŌĆō8]. Previous studies have also focused on potential associations of candidate genes with meat quality in Berkshire pigs [9ŌĆō11].
Several studies have investigated genetic factors related to meat quality in the Berkshire population using next generation sequencing and whole genome sequencing data [1,6,7,10, 11]. This technology provides a powerful approach for identifying a massive number of single nucleotide polymorphisms (SNPs) in genome-wide sequence data [12,13,]. Selective sweeps, which were inferred from whole genome sequencing data using cross-population extended haplotype homozygosity (XP-EHH) and a cross-population composite likelihood ratio method (XP-CLR), have revealed distortions in genetic patterns between Berkshire and two other maternal breeds (Yorkshire and Landrace) [1]. These selective sweeps reveal strong selection signatures in genomic regions that harbor economic trait-related genes. Therefore, the identification of positively selected genetic regions, especially in Berkshire pigs might allow the exploration of genetic mechanisms related to Berkshire-specific phenotypic traits mentioned above, including rapid and efficient muscle accumulation and desirable pork qualities etc.
Although many SNPs are phenotypically neutral, nonsynonymous SNPs (nsSNPs) that are located in protein-coding regions and cause amino-acid substitutions in corresponding protein products, can alter protein structure, stability, or function and are typically associated with disease. In human, nsSNPs cause approximately 50% mutations involved in inherited genetic diseases [14ŌĆō16]. In addition, multiple nsSNPs in innate immune genes can affect susceptibility to infection, as well as the development of inflammatory disorders and autoimmune diseases [17ŌĆō19]. The effects of nsSNPs on porcine genetic mechanisms remain under investigation.
In this study, we performed a genome-wide annotation of nsSNPs of the genes located in selective sweeps of Berkshire pigs in regard to protein function, using in silico bioinformatics tools. Notably, nsSNPs are relevant to biological functions reflective of breed characteristics [20,21]. Accordingly, we integrated and reanalyzed the whole-genome sequencing data of Berkshire, Duroc, Korean native pig, Landrace, miniature pig, and Yorkshire pigs, and wild boar, to identify genomic variants. The nsSNPs were indentified in Berkshire selective sweep regions and investigated to discover genetic mechanisms that were potentially associated with Berkshire domestication and meat quality. We further used bioinformatics tools to predict damaging amino-acid substitutions caused by novel nsSNPs of genes in Berkshire pig genome.
MATERIALS AND METHODS
Sample preparation and whole genome sequencing
The whole genome sequence data set used in this study included data of 14 Berkshire, 26 Duroc, 14 Korean native pigs, 14 Landrace (Danish), 16 miniature pigs, and 16 Yorkshire (Large White) pigs, and 10 wild boars. All data were obtained from the NCBI Sequence Read Archive database (SRP047260, SRP052927, PRJNA254936, and PRJNA281548). Especially, Berkshire, Landrace, and Yorkshire (Large White) among the 110 pigs were used in a previous study [22] to explore the evidence suggesting a genetic basis for the positive selection of meat-quality traits in Berkshire pigs.
FastQC software [23] was used to perform a quality check of raw sequence data, and Trimmomatic-0.32 [22] to remove potential adapter sequences before sequence alignment. Paired-end sequence reads were mapped to a pig reference genome (Sscrofa 10.2.75) from the Esembl database, using Bowtie2 [24] with default settings. The open-source software packages, i.e., Picard tools (https://broadinstitute.github.io/picard/), SAMtools [25], and genome analysis toolkit (GATK) [26] were used for downstream processing and variant calling. The Picard ŌĆ£CreateSequenceDictionaryŌĆØ and ŌĆ£MarkDuplicatesŌĆØ command-line tools were used to read reference FASTA sequences for writing bam files with only a sequence dictionary and to filter potential polymerase chain reaction duplicates, respectively. We used SAMtools to create index files for the reference and bam files, and then used the GATK ŌĆ£Realigner-TargetCreatorŌĆØ and ŌĆ£IndelRealignerŌĆØ arguments to perform a local realignment of sequence reads to correct misalignments caused by small insertions and deletions. Base quality scores also were recalibrated to ensure accuracy and correct machine cycle-related and sequence context-related quality variations. GATK ŌĆ£Unified GenotyperŌĆØ and ŌĆ£Select VariantsŌĆØ arguments were used to call variants according to the following filtering criteria: i) a Phred-scaled quality score of <30; ii) read depth of <5; and iii) MQ0 (total count of mapping quality zero reads across all samples Ōēź4). Phred-scaled p-values (FisherŌĆÖs exact test) >200 were filtered out to decrease false-positive calls because of strand bias. The ŌĆ£vcf-mergeŌĆØ tool in the VCF tools package [27] were used to merge all variant-calling format files for the 110 samples.
Population structure analysis
A population structure analysis was performed to infer the population structures from whole-genome sequence data of 110 pigs. The STRUCTURE program (version 2.3.4) was used to evaluate substructure among the 110 individuals from seven breeds. A Bayesian clustering analysis in this program [28,29] was used to estimate the population structure, using 65,550 nsSNPs identified from the whole-genome sequencing data of all 110 pigs. We determined that an initial burn-in of 100,000 iterations, followed by 100,000 iterations for parameter estimation, would be sufficient to ensure the convergence of parameter estimates. To identify admixtures (K parameter, STRUCTURE), we analyzed the dataset after allowing for K values of 4 and 6.
Identification of nsSNPs in Berkshire selective-sweep regions
ClueGO [30] was used to perform a gene ontology (GO) network analysis for the identified genes to infer the biological meanings of the related nsSNPs. In addition, empirical p-values were defined according to rankings to further investigate multiple nsSNP-containing genes. In the Berkshire population, genes were ranked in descending order by nsSNP number with respect to the empirical p-value ranking. In this study, ŌĆ£p-valuesŌĆØ denotes empirical p-values; here, a low p-value indicates that a locus is an outlier relative to the rest of the genome.
Prediction of damaging amino-acid substitutions in nsSNPs in Berkshire pigs
In this study, the functional effects of amino acid sequence change by nsSNPs were predicted using the following in silico algorithms: sorting intolerant from tolerant (SIFT) [31,32] and polymorphism phenotyping V2 (Polyphen-2) [33]. SIFT uses sequence homology to predict the effects of the amino-acid substitution of interest on protein functions. nsSNP-based sequence homology tests were performed to identify important amino-acid substitutions that might affect biological functions via structural modifications of proteins. In our analysis of 319 nsSNPs from Berkshire selective-sweep regions (Table 1), a SIFT score of <0.05 indicated that the SNP was deleterious and could strongly affect protein function. Moreover, a PolyPhen-2 (version 2.2.2) analysis were performed with specific empirical rules to predict the effects of amino-acid substitutions on the structures and functions of proteins [3], using amino-acid encoded by genes containing target SNPs from the Esembl database. After SIFT analysis, proteins resulting from 319 nsSNPs in Berkshire selective-sweep regions were tested using Polyphen-2. Tested nsSNPs were classified as probably damaging, possibly damaging, or benign if they received Polyphen-2 scores (range: 0 to 1) of >0.95, 0.5 to 0.95, or <0.5, respectively [33]. In this study, we expected that probably damaging and possibly damaging SNPs would affect protein functions. The results from our analyses of the effects of nsSNPs on protein function are shown in Tables 1, 2.
RESULTS
DNA sequencing data preprocess and genetic variant calling
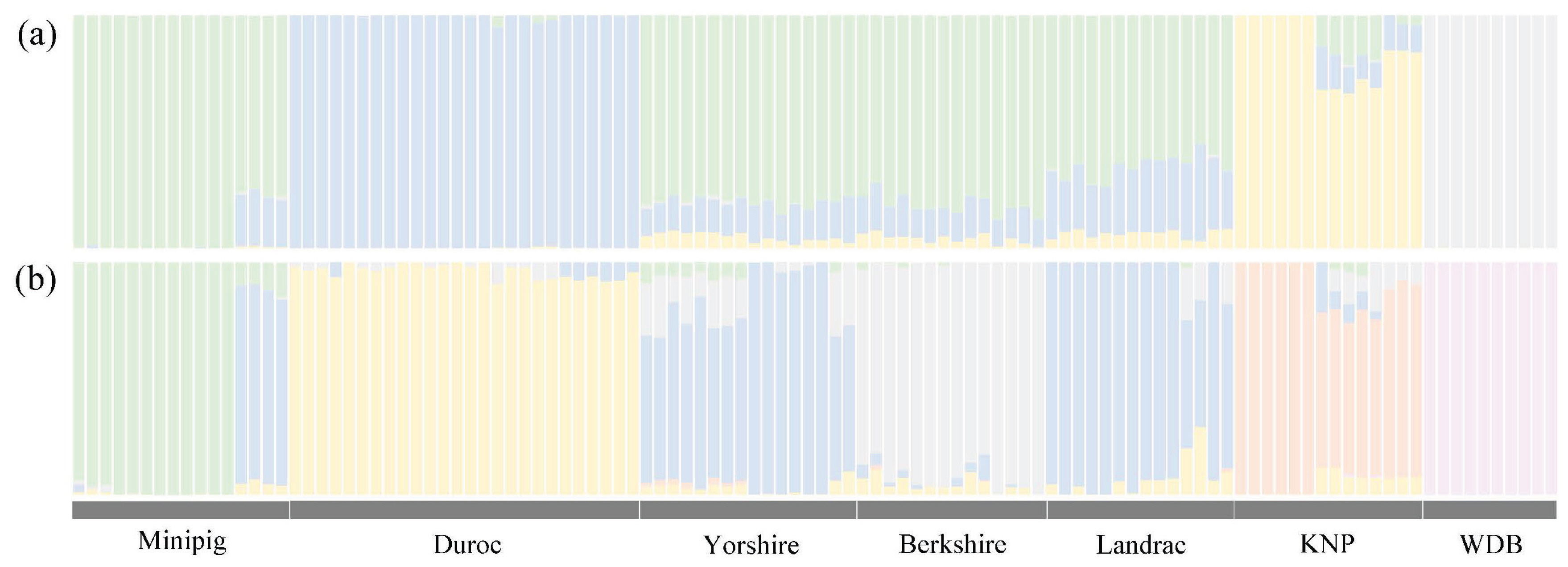
After excluding tri-allelic SNPs (283,097 SNPs; 0.91% of 31,113,755 total SNPs), a total of 30,830,658 SNPs were extracted from the whole-genome sequences of 110 pigs, including 14 Berkshire individuals (Supplementary Table S1), and annotated all extracted SNPs using SnpEff version 4.1a [34]. Subsequently, all SNPs were divided into 19 functional classes, including nsSNPs (Supplementary Figure S1). Most SNPs were located in intergenic or intron regions, and 65,550 nsSNPs (0.223% of total SNPs) were identified in 12,469 protein-coding genes from all SNPs in genic regions (except splice regions). Of these nsSNPs, SNP frequency of 26,644 were not zero, and 8,177 genes in Berkshire population contained two or more SNPs. A population structure analysis was performed based on genotypic information to evaluate the genetic relationships among breeds, and found that Landrace and Yorkshire pig breeds and miniature pigs were clustered into one group and were distinct from Duroc pigs, Korean native pigs, and wild boars (K = 4 in Figure 1). However, Berkshire populations were distinct from Yorkshire, Landrace, and miniature pigs at K = 6 (Figure 1).
Identification of nsSNPs in Berkshire selective-sweep regions
In this study, 155 genes (56 genes from XP-EHH plus 114 genes from XP-CLR) were identified in selective-sweep regions of the Berkshire genome. Furthermore, 319 of the total 65,550 nsSNPs belonged to 73 of 155 genes in Berkshire selective sweep regions (Supplementary Table S2). Genotype information for these 319 SNPs in Berkshire, Landrace, and Yorkshire populations is presented in Figure 2. In addition, by defining empirical p-values according to rankings to further investigate multiple nsSNP-containing genes, and observed 12,469 genes in our 110-pig population contained multiple SNPs. In the Berkshire population, we ranked genes in descending order by nsSNP number with respect to the empirical p-value ranking. The p-values were assigned to 12,469 genes; those with p-values of <0.05 (5%) as were thought to contain significantly large numbers of nsSNP and were assumed to be strongly associated with Berkshire characteristics. Thirteen of 73 genes with nsSNPs in the Berkshire selective-sweep region had p-values of <0.05 (Supplementary Table S2).
nsSNPs in the Berkshire selective-sweep region
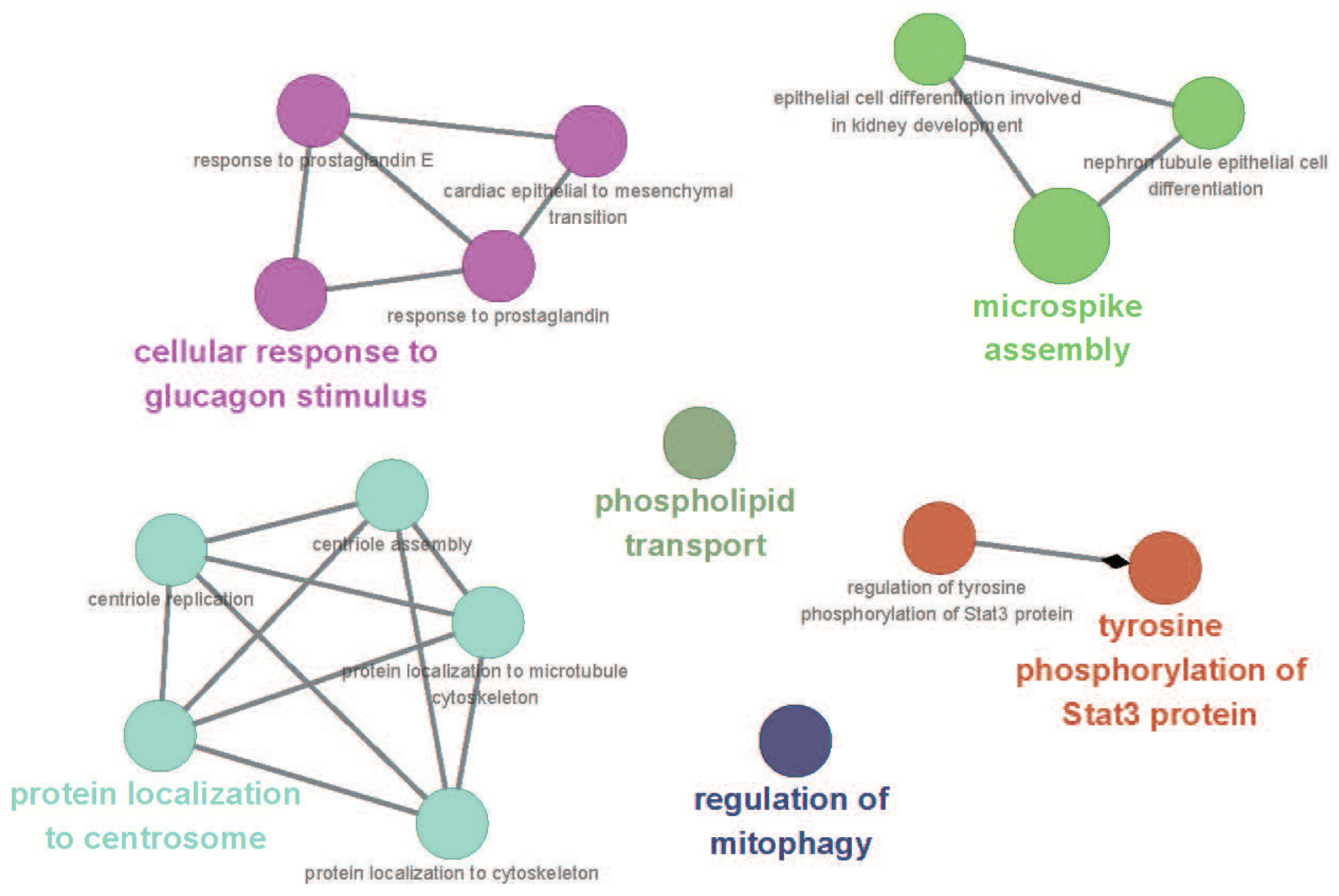
Detailed information of genes that were analyzed in this study is presented in Supplementary Table S2, and the findings of a GO network analysis for 73 genes are presented in Figure 3 and Supplementary Table S3. Through our GO network analysis, 17 of 73 genes, i.e., ATP binding cassette subfamily A member 13 (ABCA13), actin alpha2 (ACTN2), ATPase phospholipid transporting 9A (ATP9A), C2 calcium dependent domain containing 3 (C2CD3), centrosomal protein 152 (CEP152), growth hormone (GH), glucagon like peptide 2 receptor (GLP2R), GNAS complex locus (GNAS), interleukin 22 receptor subunit alpha 2 (IL22RA2), microtubule associated protein 1 light chain 3 beta (MAP1LC3B), I-BAR domain containing (MTSS1), olfactomedin 1 (OLFM1), slowmo homolog 2 (SLMO2), prominin 1 (PROM1), sperm associated antigen 5 (SPAG5), transforming growth factor beta receptor 3 (TGFBR3), ubiquitin specific peptidase 36 (USP36), were found associated with six major GO terms. TGFBR3 and GLP2R, which have been previously reported as major candidate meat-quality genes according to the signatures of selection scan of the whole genome sequences in Berkshire pigs [1], were closely related to the cellular response to glucagon stimulus, one of six significant GO terms (Supplementary Table S3). In the previous study [35], a SNP in the intron region of GLP2R associated significantly with the intramuscular fat in a Large White├ŚMinzhu intercross population. Moreover, we detected a novel nsSNP (Chr12:574,612,64) in the exon region of GLP2R [35]. In general, glucagon induces lipolysis in mammals under conditions of insulin suppression, and we considered that amino-acid substitutions caused by nsSNPs in the Berkshire genome might affect lipolysis, thus affecting the high proportions of neutral lipid fatty acids and marbling fat associated with this breed. TGFBR3 plays important roles in muscle and adipose tissue development [36], and increased transcription of TGFBR3 mRNAs was observed in pigs with high lipid deposition and composition. We therefore assumed that four genes (GNAS, OLFM1, GLP2R, TGFBR3) related to cellular responses to glucagon stimulus would be affected by selective pressure over time, to establish characteristics of Berkshire that have been selected artificially.
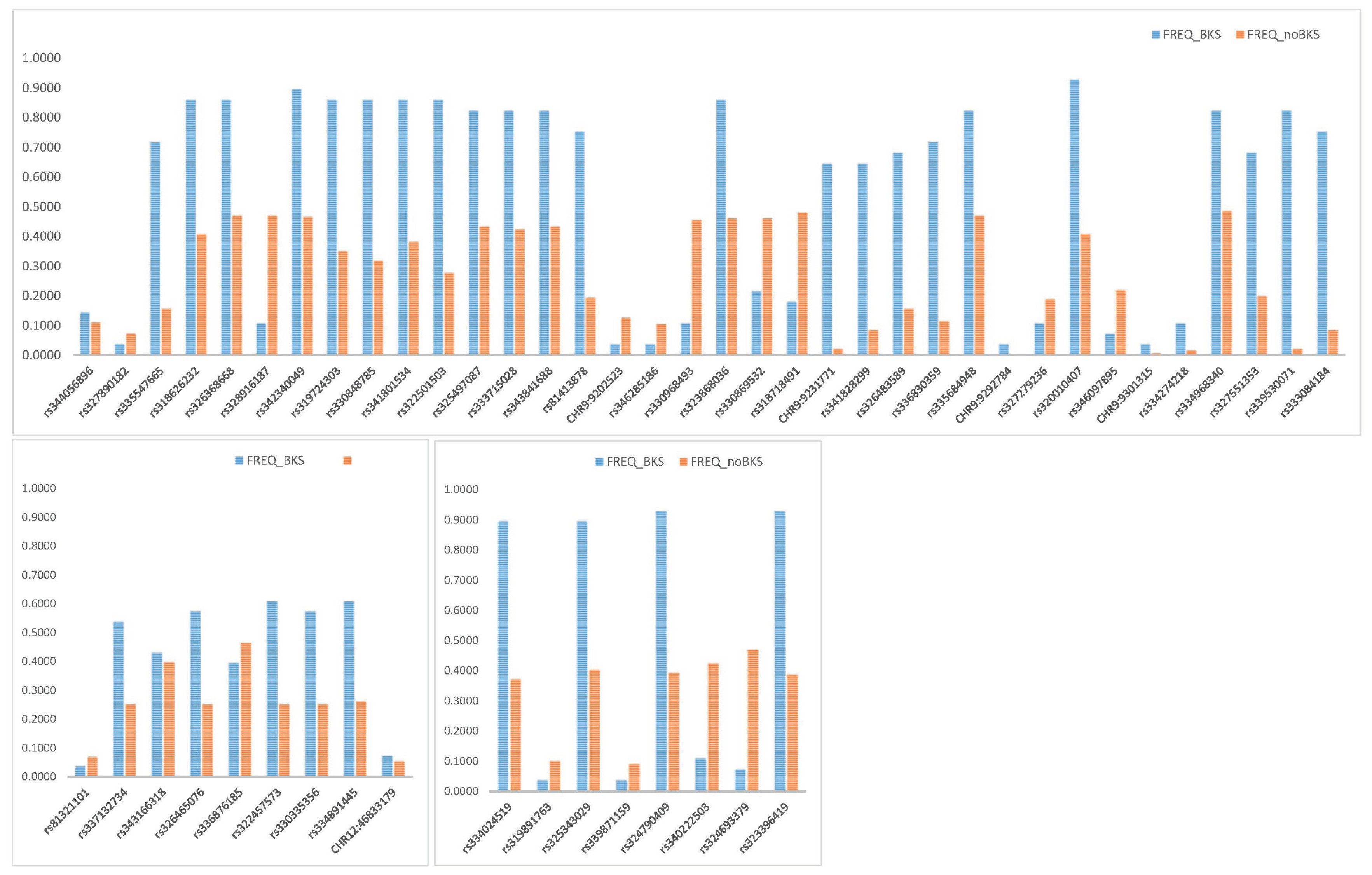
Three genes (C2CD3, CEP152, SPAG5) were associated with protein localization to the centrosome, cytoskeleton, and microtubule cytoskeleton (Figure 3). Rutkowski et al [37] reported that the coalescing mechanism associated with large lipid droplet formation was microtubule-dependent, and therefore disruption of cytoskeletal remodeling would affect lipid droplet maintenance and dynamics during lipolysis. Furthermore, dynamic changes in adipocyte size (expansion and reduction) are thus critically dependent on the environments [37]. Our comparison of SNP frequencies in C2CD3, CEP152, and SPAG5 led us to consider that the high levels of genetic variation might be linked to the high lipid deposition in Berkshire pigs (Figure 4). ABCA13, ATP9A, and PRELID3B are involved in phospholipid transport. Notably, the fatty acids of phospholipids are particularly important in developing pork flavor, and obvious genetic effects have been observed on the total fatty-acid concentrations in muscle tissues. Another study observed higher phospholipid concentrations in the longissimus and psoas muscles of Berkshire purebred pigs compared to Large White pigs [38]. Once engaged with a specific receptor or ligand, GH and IL22RA2 can transmit signals via tyrosine-phosphorylated STAT3 to regulate the transcriptional activities of lipid metabolism-related genes [39].
We ranked genes by empirical p-values based on number of nsSNP and number of nsSNPs among the population of 110 pigs. Overall, 715 of 12,469 genes had p-values of <0.05 and contained at least eight SNPs. We assumed that these 715 genes would be significantly affected in Berkshire pigs; in other words, those genes would be strongly associated with Berkshire domestication and characteristic establishment. Among these 715 genes, we identified 13 nsSNP-containing genes with significant p-value-based ranks in the Berkshire selective-sweep region. The highest rank gene was C2CD3 among 13 genes, which encodes a protein that regulates centriole elongation. In this study, C2CD3, along with CEP152 and SPAG5, was associated with protein localization to the centrosome, cytoskeleton, and microtubule cytoskeleton in the GO network analysis. Interestingly, CEP152 (p-value = 0.045) and SPAG5 (p-value = 0.036) were also included among the 13 significant genes. Details of the nsSNP analysis results are summarized in Supplementary Figure S1.
SIFT analysis of nsSNPs in the Berkshire selective-sweep region
According to the SIFT analysis, 49 of 319 nsSNPs were classified as deleterious (some were deleterious with low confidence). PolyPhen-2 was used to calculate the true-positive rate as a fraction of predicted mutations (Figure 1), and 58 nsSNP-related amino-acid variants (sum of possibly damaging and probably damaging in Table 1) in the Berkshire selective-sweep region may cause deleterious functional effects on the associated genes; these findings overlapped with SIFT results for 27 nsSNPs. Therefore, it is reasonable to assume that these 27 SNPs may have a considerable impact on biological mechanisms during Berkshire domestication (Table 1). Thyroglobulin (TG), coiled-coil domain-containing protein 30 (CCDC30), and ribosomal RNA processing 1B (RRP1B) each contained multiple SNPs and have strong effects on protein stabilization (Table 2). In particular, the ranked p-value of TG and CCDC30 were 0.05 or less. CCDC30 contained 25 nsSNPs in the Berkshire population; of these, eight SNPs, i.e., rs331099959, rs3214 01589, rs330010762, rs342319002, rs335124185, rs323439800 and 2 novel SNPs, were identified to have considerable effects in both the SIFT and PolyPhen-2 analyses. However, the detailed physiological mechanism of CCDC30 in the Berkshire domestication process remains to be studied. TG plays an important role in adipocyte growth and differentiation. In this study, among the 17 nsSNPs of TG in Berkshire pigs, 4 nsSNPs were identified to have considerable impacts on protein stabilization. Notably, a previous study identified TG as a major candidate genes related to meat quality via Berkshire positive selection scans (XP-EHH and XP-CLR) [1]. In the Berkshire population, RRP1B, which encodes a ribosomal RNA processing protein homolog, contained two nsSNPs with potentially strong effects on protein residue stabilization. Previously, Grant et al [40] demonstrated the differential transcription of RRP1B in lean and obese individuals [40], which led us to presume that RRP1B may be strongly correlated with meat quality in domesticated Berkshire pigs. Although C2CD3 contained the highest number of Berkshire nsSNPs among the 12,469 evaluated genes, only one of the 36 SNPs was thought to have a strong effect on protein function. Like C2CD3, CEP152, and SPAG5 each had ranking p-values of <0.05 but there is only one SNP thought to cause a significant amino-acid substitution (Table 2). Similarly, otogelin like (OTOGL), seizure threshold 2 (SZT2), family with sequence similarity 208 member B (FAM208B), and integrin subunit alpha 1 (ITGA1) each had ranking p-values of <0.05 but only one SNP expected to affect protein function.
DISCUSSION
Regarding the interest of the meat-production industry in quality improvement, a previous study performed a selective sweep-based genetic investigation of Berkshire pigs to determine information essential to domestic breeding [1]. Various SNPs were used in these selective sweeps. A nonsynonymous substitution of amino acid by a nsSNP may result in biological changes and are therefore subject to natural selection. An evaluation of positive selection throughout the Berkshire whole genome sequence data indicated the need for an advanced, nsSNP-based interpretation. In this study, we aimed to predict and interpret the impacts of amino acid substitutions by nsSNPs of Berkshire pigs by in silico tools, under the assumption that these elements might associate with important and previously unrevealed biological mechanisms related to Berkshire characteristics. Our deep analyses identified 319 nsSNPs in the Berkshire selective sweep regions that were subjected to a GO network analysis. Here, the SNPs were classified according to six major GO terms, most of which were associated with process involved in lipid metabolism.
We investigated the nsSNPs of seven major meat-quality candidate genes, i.e., TG, fatty acid binding protein 1, Akirin 2, TGFBR3, junctophilin 3, intercellular adhesion molecule 2, and endoplasmic reticulum to nucleus signaling 1, detected via positive selection scans in a previous study [1]. In the Berkshire population, three of the genes did not contain nsSNPs; of the remaining four, all genes contained at least one nsSNP, and JPG3 contained three nsSNPs. Only TG contained 17 nsSNPs. Accordingly, we searched additional genes that have a potential impact on Berkshire domestication. First, we identified several genes associated with various biological processes associated with lipid metabolism (e.g., cellular responses to glucagon stimulus, protein localization to centrosome, and phospholipid transport and tyrosine phosphorylation of STAT3 protein) through a GO network analysis (Figure 3; Supplementary Table S3). Second, we conducted a differential search for genes that were enriched for nsSNPs specifically in the Berkshire population. Each nsSNP has been assigned a ranking based on p-values and identified 13 genes that met our criterion (p-value <0.05) (Supplementary Table S2). Of these, interestingly, only TG overlapped with previously detected major meat-quality candidate genes [1]. Furthermore, all three genes (C2CD3, CEP152, SPAG5) associated with protein localization to the centrosome in our GO ontology were included among this 13-gene set, and contained more nsSNPs than others in the Berkshire selective-sweep region. Surprisingly, most SNPs in these three genes were present at very high frequencies in the Berkshire population compared to other pig breeds. C2CD3 contained 36 nsSNPs, the highest number in the Berkshire population, and most were present at high allele frequencies. We expect that the variants in these three genes likely played a crucial role in Berkshire domestication and therefore warrant additional studies in the future. Third, we used SIFT [32] and PolyPhen-2 [33] to predict the possible effects of the 319 nsSNPs in Berkshire selective-sweep regions on protein function. Twenty-seven SNPs were predicted associated with biological functions at the protein levels. These results suggest that genes related to these 27 SNPs were associated with Berkshire meat quality.
Our results strongly suggest that genetic variants in Berkshire pigs are closely associated with important meat-quality factors such as increased lipid deposition and composition. Our study is distinct from others because of its use of nsSNPs to evaluate functional impacts of amino acid change caused by nsSNPs in the Berkshire genome. Nonetheless, this study has limitations; first, even various bioinformatics tools were adapted for the analyses but information that we studied in this study come from human variants database as there is no livestock specific database to evaluate functional impact of nsSNPs so far. Also, this study lacks experimental validation on the functionality of nonsynonymous amino acid substitution that is predicted by in silico analyses. Further study warrants experimental validation to prove the functionality of amino acid substitutions by nsSNPs of genes found in Berkshire pigs, which will allow us to identify causal variations to the traits of interest and ultimately to use variants as genetic markers.
Taken together, our study allowed an investigation of novel biological facts that could promote a better understanding of Berkshire domestication, and some of our results overlapped with and corroborated those of previous studies. We expected that many of our results will provide deep insights into the genetic factors underlying Berkshire domestication.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







