Genome-wide association study reveals genetic loci and candidate genes for average daily gain in Duroc pigs
Article information

Abstract
Objective
Average daily gain (ADG) is an important target trait of pig breeding programs. We aimed to identify single nucleotide polymorphisms (SNPs) and genomic regions that are associated with ADG in the Duroc pig population.
Methods
We performed a genome-wide association study involving 390 Duroc boars and by using the PorcineSNP60K Beadchip and two linear models.
Results
After quality control, we detected 3,5971 SNPs, which included seven SNPs that are significantly associated with the ADG of pigs. We identified six quantitative trait loci (QTL) regions for ADG. These QTLs included four previously reported QTLs on Sus scrofa chromosome (SSC) 1, SSC5, SSC9, and SSC13, as well as two novel QTLs on SSC6 and SSC16. In addition, we selected six candidate genes (general transcription factor 3C polypeptide 5, high mobility group AT-hook 2, nicotinamide phosphoribosyltransferase, oligodendrocyte transcription factor 1, pleckstrin homology and RhoGEF domain containing G4B, and ENSSSCG00000031548) associated with ADG on the basis of their physiological roles and positional information. These candidate genes are involved in skeletal muscle cell differentiation, diet-induced obesity, and nervous system development.
Conclusion
This study contributes to the identification of the casual mutation that underlies QTLs associated with ADG and to future pig breeding programs based on marker-assisted selection. Further studies are needed to elucidate the role of the identified candidate genes in the physiological processes involved in ADG regulation.
INTRODUCTION
The pig (Sus scrofa) is an economically important farm animal, and the Duroc×(Landrace× Yorkshire) three-way cross hybrid (DLY) pig is the most popular commercial pig breed worldwide. The growth performance of DLY pigs is directly affected by that of their terminal male parent, the Duroc pig. Growth performance in commercial swine productions systems is measured on the basis of numerous economically important production traits, among which the average daily gain (ADG) is the most important. Pigs with higher ADG can achieve the target market weight within a shorter period than those with lower ADG, thus saving considerable feeding costs. Therefore, pig farmers have aimed to increase the ADG of swine to improve profitability. Fortunately, molecular technologies have helped improve the ADG of commercial swine. The association of several major genes, such as translation elongation factor 1 and corticotropin releasing hormone, and quantitative trait loci (QTLs) with ADG has been confirmed; breeding on the basis of these genes and QTLs might result in the considerable improvement of the ADG of commercial pig herds in the near future [1–4].
Previous studies have utilized microsatellite markers to identify the QTLs or major functional genes of quantitative traits, such as ADG [2,5]. These QTLs, however, are based on linkage analyses and map construction with few genetic markers, resulting in large QTL spans. QTL spans could be shortened and novel associated variants could be identified via genome-wide association (GWA) studies through the use of recently developed high-throughput commercial genotyping platforms and single nucleotide polymorphisms (SNPs) [6,7]. Thousands of QTLs that control a wide range of pig traits have been identified, and 563 QTLs for ADG have been deposited in PigQTLdb (http://www.animalgenome.org/cgi-bin/QTLdb/SS/summary). Nevertheless, only a handful of causative genes have been identified via QTL mapping analysis because QTLs span large chromosomal regions that, in turn, comprise hundreds of genes [8]. Therefore, minimizing the QTL region would facilitate the discovery of causative genes.
GWA is a powerful strategy for the genetic dissection of trait loci in humans and other animals [9,10]. In this method, high-density molecular markers screened from genomes are used to analyze the association between phenotypic traits and marker data. GWA was first proposed by Risch et al [11]. More than 60,000 SNPs can be analyzed with high efficiency following the development of high-throughput commercial genotyping platforms for pigs [12]. Consequently, this development has enabled the realistic estimation of genetic diversity and genome structure [13]. In addition, more than one model can be used for GWA analysis [14,15].
In this study, we subjected the genomic data of 390 Duroc boars to GWA. We aimed to analyze population-based associations and identify SNPs and QTLs associated with the ADG of pigs. We utilized Illumina Porcine SNP60 BeadChip as the genotyping platform and the mixed linear model (MLM) and multi-locus random-SNP-effect mixed linear model (mrMLM) for GWA [16].
MATERIALS AND METHODS
Ethic statement
The experimental procedures used in this study met the guidelines of the Animal Care and Use Committee of the South China Agricultural University (SCAU) (Guangzhou, People’s Republic of China). All animal experiments in this study were approved by the Animal Care and Use Committee of the SCAU with the approval number SCAU#0017.
Animals and phenotypes
A total of 390 male Duroc pigs were procured from Guangdong Wen’s Foodstuffs Co., Ltd. (Guangdong, China). The pigs were born over the period of 2011 to 2014. They were randomly assigned to groups and housed in half-open cement-floor pens. Each pen contained 10 animals, with an average floor space of 2 m2 per pig. The ADG phenotype was identified using Osborne FIRE Pig Performance Testing System (Kansas, NE, USA) by the personnel of Guangdong Wen’s Foodstuffs Group Co., Ltd. (Guangdong, China) when the pigs weighed from 30 kg to 100 kg. All individuals were fed under the same standard conditions with the same feedstuff. Ear tissue was collected as follows: the pig’s ear was first cleaned with 75% alcohol. Then, a clear forfex was used to cut out a small fraction of ear tissue. The wound was then treated with tincture of iodine. The protocol for ear tissue collection was approved by the ethics committee of SCAU.
DNA isolation, genotyping, and quality control
DNA was extracted from each sample of ear tissue following the standard phenol/chloroform extraction method. All DNA samples were qualified and normalized to a final concentration of 50 ng/μL. DNA quality was assessed on the basis of the ratios of light absorption (A260/280 and A260/230) and electrophoresis. The quality and concentration of each genomic DNA sample met the requirements for the Illumina SNP genotyping platform. The 390 individuals were genotyped using the porcine SNP60K Beadchip of Illumina (San Diego, CA, USA) [12]. Quality control was performed using PLINK v 1.07 software [17]. SNP markers with genotype missing rates >0.05, call rate <95%, minor allele frequencies <0.01, and Hardy–Weinberg p≤10E-06 were excluded. Unmapped SNPS and SNPs located on sex chromosomes were removed in accordance with the Sus scrofa10.2 assembly of the reference genome [18]. Samples and SNPs that passed the filter were selected for subsequent GWA analysis.
Generalized mixed linear model and population stratification assay
Association analyses based on the generalized MLM were conducted using GenABEL in R software [15]. This model included a random polygenic effect, and the variance–covariance matrix was proportionate to genome-wide identity-by-state [19]. The formula of the model is given by the mathematical expression Y~μ+Xb+Kw+Sc+Za+e. In this expression, Y is the vector of phenotypes; μ is the overall mean; b is the vector of fixed effects, including yearly and seasonal effects; w is the vector of the body weight of individuals and is considered as a covariate; and c is the vector of SNP effects. In addition, a is the vector of random additive genetic effects with a~ N(0, Gσα2), where G is the genomic relationship matrix calculated from the corrected pedigree and σα2 is the polygenetic additive variance. Finally, K is the regression coefficient of slaughter weight, and e is the vector of residual errors with e~N(0, Iσe2), where I is the identity matrix and σe2 is the residual variance. X, S, and Z are incidence matrices for b, c, and a, respectively.
Significant thresholds were identified through the Bonferroni method, in which the conventional p-value was divided by the number of performed tests [20]. A SNP was considered to have stringent genome-wide significance at p< 0.05/N and suggestive significance at p< 1/N, where N stands for the number of SNPs tested in the analyses. In this study, the significant and suggestive thresholds were 1.20E−6 (0.05/35971) and 2.40E−5 (1/35971), respectively.
Population stratification can affect the reliability of GWA results. The quantile–quantile (Q–Q) plot is a suitable tool for assessing the presence of population stratification. It is used to examine the distribution of test statistics generated from thousands of association tests and assessing their deviation from the null distribution (the distribution expected under the null hypothesis of no SNP associated with the trait) [21]. In the Q–Q plot (Figure 1A), the horizontal axis and the vertical axis represent the expected −log10P and the observed −log10P, respectively. The diagonal line represents y = x, and the shaded region represents the 95% confidence interval based on β-distribution. Overall deviation above the diagonal identity line may indicate severe population stratification [22]. Deviations from the diagonal line suggest that either the assumed distribution is incorrect or that the sample contains values that arose in some other manner, as in a true association [23]. In the present study, the Q–Q plot was generated using R software.

Quantile–quantile and Manhattan plots. (A) Quantile–quantile (Q–Q) plots showing the observed versus expected log p-values for the average daily gain (ADG) trait. The horizontal axis indicates the expected 2log10 (p-values) and the vertical axis indicates the observed 2log10 (p-values). The diagonal line represents y = x, which corresponds to the null hypothesis, and the shaded region shows 95% confidence interval based on β-distribution. (B) Manhattan plot showing the significance of association between 35,791 single nucleotide polymorphisms (SNPs) and the ADG trait. In the Manhattan plots, the negative log10 p-values of the quantified SNPs were plotted against their genomic positions. The solid and dashed lines indicate the 5% genome-wide and chromosome-wide Bonferroni-corrected thresholds, respectively.
Haplotype or linkage disequilibrium (LD) block analyses were performed for chromosomal regions with multiple significant SNPs clustered around the peak SNP. PLINK v1.07 and Haploview version 4.2 were used to complete haplotype blocks. Then, two candidate genes that were located in the regions of the haplotype block were selected in accordance with functional information retrieved from the Ensembl database (www.ensembl.org) [24,25].
Multi-locus random-SNP-effect mixed linear model
Common GWA methods are based on single marker analysis and fixed-SNP-effect MLM [14]. These methods are limited by the over-strict correction (Bonferroni correction) of multiple tests. Thus, the genotypic information of potential SNP loci cannot be efficiently used. The mrMLM treats the effect of SNPs as random and allows the modified Bonferroni correction to be used for calculating the threshold p-value for significance tests [16]. GWA based on mrMLM was employed using the R package “mrMLM.” We set the significant threshold of log of odds (LOD) score as 4 in our study in accordance with that in Wang et al [26].
RESULTS
Summary of phenotypic and SNP data
Table 1 shows the phenotype values of the ADG of experimental pigs. The data in the table include the number of individuals, as well as the mean, standard deviation, maximum, and minimum ADG values. After filtering for quality control, 750 SNPs on sex chromosomes, 5,252 unmapped SNPs, 840 markers that failed the HWE test, 489 SNPs that failed the missingness test, and 18,792 SNPs that failed the frequency test were excluded. A total of 35,971 SNPs (35,971/61,565) were used for subsequent analysis (Supplementary Table S1). Supplementary Table S1 shows that the average marker density (physical distance) of adjacent SNPs on the same chromosome was approximately 0.06 Mb and ranged from 0.05 Mb (Sus scrofa chromosome [SSC]14) to 0.08 Mb (SSC1).

Description of ADG of the studied Duroc population based on phenotype statistics
Assessment of population stratification
Population stratification is a major threat to the reliability of the GWA results [21]. The Q–Q plots of test statistics in the GWA are shown in Figure 1A. The plot reveals that the ADG data lack clear overall systematic bias. In this study, the average genomic inflation factor (λ) of GWA for ADG was 1.03, suggesting the absence of population stratification.
Genome-wide association study
GWA results through generalized MLM are illustrated in Figure 1B and Table 2. The p-values of (in terms of –log10P) the profiles of all SNPs tested for association with the ADG trait are shown in Figure 1B. Table 2 shows the genome-wide and chromosome-wide significant SNPs for ADG. Only one genome-wide significant (significant) SNP (MARC0025058) and two chromosome-wide significant (suggestive) SNPs (MARC 0074154, H3GA0016186) were identified. Then, two significant SNPs (MARC0025058, H3GA0016186) were mapped to two haplotype blocks spanning 433 and 774 Kb (Figure 2A). Information about Sus scrofa10.2 retrieved from the Ensembl database revealed that 12 genes (DEAD-box helicase 31, general transcription factor 3C subunit 4, ENSSSCG00000005732, ENSSSCG00000005733, ENSSSCG00000005735, ral guanine nucleotide dissociation stimulator, carboxyl ester lipase, general transcription factor 3C subunit 5 [GTF3C5], ENSSSCG 00000027595, ENSSSCG00000022250, ENSSSCG00000023474, and GFI1B) are located in a block on SSC1. Eight genes, including high mobility group AT-hook 2 (HMGA2), LLP homolog, long-term synaptic facilitation, ENSSSCG00000000473, ENSS SCG00000000474, interleukin 1 receptor associated kinase 3, ENSSSCG00000000476, DNA helicase B, and ENSSSCG0000 0018696, were identified in a block on SSC5 after further analysis. The functional and locational information of the identified genes were retrieved from the Ensembl database and showed that in the haplotype blocks, GTF3C5 and HMGA2 stood out as candidate genes that may be associated with ADG. Through the pig genome database, we found that marker MARC0025058 is on the seventh intron of GTF3C5 on SSC1. Marker H3GA 0016186 is located 78,423 bp upstream of HMGA2 on SSC5. The location of other genes in haplotype blocks are shown in Supplementary Table S2.
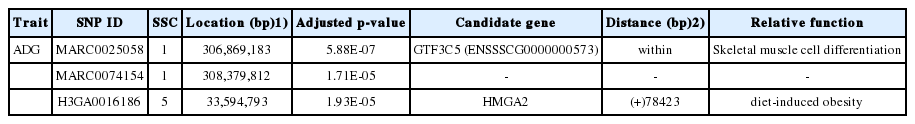
Location of significant SNPs and candidate genes for ADG

Linkage disequilibrium blocks (LD) on Sus scrofa chromosome (SSC) 1 and SSC5. (A) Linkage disequilibrium blocks on SSC1. (B) Linkage disequilibrium blocks on SSC5. LD blocks are marked with triangles. Values in boxes are the LD (r2) between SNP pairs. The boxes are colored in accordance with the standard Haploview color scheme: LOD>2 and D′ = 1, red; LOD<2 and D′<1, white (LOD is the log of the likelihood odds ratio, a measure of confidence in the value of D′).
GWA results obtained through mrMLM are illustrated in Supplementary Figure S1 and Table S3. All significant SNPs with LOD scores greater than four are shown in Supplementary Figure S1, and the detailed information of these SNPs are shown in Supplementary Table S3. DRGA0006936, DRGA000 9666, INRA0041583, and ASGA0074790 were found on SSC 6, SSC 9, SSC 13, and SSC 16, respectively, and are all associated with ADG. Six candidate genes (GTF3C5, HMGA2, nicotinamide phosphoribosyltransferase [NAMPT], oligodendrocyte transcription factor 1 [OLIG1], pleckstrin homology and Rho GEF domain containing G4B [PLEKHG4B], and ENSSSCG 00000031548) were selected based on the information from the Sus scrofa 10.2 database of Ensembl.
DISCUSSION
Duroc population and GWA
Duroc pigs can be terminal sire of DLY, which is the most popular commercial pig breed. Improving the growth performance traits, such as ADG, of Duroc is crucial for the pig industry. Thus, identifying the QTL and major genes responsible for the ADG of Duroc is necessary to facilitate the future molecular breeding of commercial pig breeds. GWA provides an efficient method to search for ADG-related genes in pigs. GWA studies are commonly based on single marker analysis and fixed-SNP-effect MLM [14]. These methods, however, are limited by the over-strict Bonferroni correction of multiple tests. Thus, the genotypic information of potential SNP loci cannot be efficiently used. In this study, the mrMLM was employed to estimate the association results produced by MLM based on single-SNP tests and to efficiently use the phenotypic and genotypic information obtained from an enormous amount of accessions and SNPs. Results showed that the two GWA models have the same effectiveness for discovering significant ADG-associated SNPs. However, the GWA based on mrMLM discovered additional SNPs that potentially affect the performance of ADG.
Comparison of QTLs identified in this study with previous studies
Since Risch et al [11] first proposed the concept of GWA in 1996, it has been a useful method for investigating major genes or diminishing the regions of QTLs associated with specific phenotypes. We utilized two methods to perform the GWA of 390 Duroc individuals. We identified seven SNPs that are significantly associated with the ADG of pigs. Five SNPs on SSC1, SSC5, SSC9, and SSC13 are located in previously reported QTL regions that are associated with the ADG of pigs [27–30]. In the same QTL region of SSC1, Meng et al [31] and Fontanesi et al [32] discovered the H3GA0004299 and ALGA 0009614 SNPs that are associated with the ADG of American Yorkshire pigs and Large Italian White pigs, respectively. In the same QTL region of SSC5, Fontanesi et al [32] discovered four SNPs (M1GA0007246, M1GA0007255, M1GA0007258, and M1GA0007286) that are associated with ADG. The results of previous studies suggested that the QTL associated with ADG might have similar locations in different pig breeds. Howard et al [33], however, found that the QTL span from 176.19 Mb to 177.76 Mb on SSC1 and that from 84.01 Mb to 84.74 Mb on SSC5 are associated with ADG in Duroc boars. This result is consistent with previous reports that genes for quantitative traits, such as ADG, are distributed on numerous chromosomal regions of pigs [34]. Moreover, we found two other SNPs DRGA0006936 on SSC6 and ASGA0074790 on SSC16 that have not been included in any previously reported QTLs. The nearest genes of these two SNPs are ENSSSCG00 000031548 and PLEKHG4B. Although the functions of the ENSSSCG00000031548 and PLEKHG4B genes are poorly elucidated, previous studies have reported that the latter is associated with the development of the human brain and nervous system [35]. Results indicated that two QTLs might be associated with the ADG trait in the pig population.
Candidate genes
In this study, seven SNPs that are significantly associated with the ADG trait were detected on SSC1, SSC5, SSC6, SSC9, SSC13, and SSC16. Six candidate genes that are located close to significant or suggestive SNPs were considered as important candidate genes on the basis of functional and locational information. Functional gene analysis revealed that these candidate genes are mainly involved in skeletal muscle cell differentiation, diet-induced obesity and nervous system development. This result is consistent with that of a previous study [36]. After alignment the sequence of the GTF3C5 gene (http://blast.ncbi.nlm.nih.gov/Blast.cgi), we found that swine GTF3C5 exhibited over 85% and 81% homology with human and mouse GTF3C5, respectively. This result indicated that the functions of GTF3C5 in these three species are highly similar. Additionally, information from the UniProt database (http://www.uniprot.org/uniprot/F1S0S0) revealed that the GTF3C5 gene is indispensable in skeletal muscle cell differentiation in pigs. In humans, the GTF3C5 gene mediates the general protein transcription factor 3C polypeptide 5 [37,38]. The function of GTF3C5 has been studied in mice, and results showed that individuals that are homozygous for GTF3C5 are abnormal and those that are recessive for the gene are non-viable [39]. This result suggested that this gene has crucial roles in embryonic development and in postnatal growth and development. Thus, functional studies on this gene are necessary.
The HMGA2 gene encodes small, chromatin-associated protein high-mobility group AT-hook 2, which belongs to the non-histone chromosomal high-mobility group A family of DNA-binding proteins [40]. This protein can modulate transcription and promote or inhibit the action of transcriptional enhancers by altering chromatin structure or by facilitating the assembly of the multiprotein complexes of transcriptional factors [41–43]. Previous studies have revealed that HMGA2 is highly expressed during embryogenesis; this expression pattern indicates its crucial role in growth and development [44–46]. Knocking out the mouse counterpart of HMGA2 demonstrated that this gene is involved in diet-induced obesity [47]. Given that murine and porcine HMGA2 genes are highly homologous, HMGA2 may have a similar function associated with ADG in pigs. Furthermore, previous studies have confirmed that HMGA2 is overexpressed in malignant and benign tumors and is associated with certain characteristic cancer-promoting mutations [48,49]. Notably, HMGA2 is related to height [50,51]. All of the above described functions are indirectly or directly related to weight gain. The majority of functional studies on HMGA2 have focused on its role in human or mouse systems. Additional studies on porcine HMGA2 should be conducted in the future, not only because this gene is related to weight gain, height, carcinogenesis, and tumorigenesis, but also because pigs can be useful animal models for human growth or disease.
Studies on human and mouse systems have shown that NAMPT genes can encode the enzyme nicotinamide 5-phosphoribosyl-1-pyrophosphate transferase, which catalyzes the rate-limiting step in nicotinamide adenine dinucleotide biosynthesis, and that NAMPT is preferentially secreted by visceral fat tissue [52]. Cepica et al [53] found that this gene is associated with the muscling, growth, fat deposition, and fat-to-meat ratio of pigs in the wild boar×Meishan F2 family. In combination with our findings, this gene might be an important candidate for molecular breeding studies that aim to improve the ADG of pigs. OLIG1 and PLEKHG4B genes are associated with the development of the human brain and nervous system and may also encode for neurotropic factors [35,54].
Future studies are necessary to understand or prove the mechanism that underlies the effects of the selected candidate genes on ADG. Future research could include gene sequencing and the identification of mutations, additional statistical association testing, and cell experiments to allow the comparison of differences or associations between mutant and normal cell lines.
CONCLUSION
The present GWA study was based on two methods and identified seven SNPs that are significantly associated with the ADG of pigs. Six QTL regions for ADG were identified, including four previously identified QTLs on SSC 1, SSC5, SSC9, and SSC13 and two novel QTLs on SSC6 and SSC16. Six candidate genes were singled out on the basis of their functional annotations, positions, and expression patterns in related tissues. The present findings will contribute to the further identification of the casual mutation underlying these QTLs and the future improvement of ADG by pig breeding programs.
Supplementary Data
ACKNOWLEDGMENTS
This work was supported by grants from the Science and Technology Planning Project of Guangdong Province (2015B020 231010, 2017B020201012), the Natural Science Foundation of China (31601912), and the Natural Science Foundation of Guangdong Province (2016A030310447). The authors are thankful to Guangdong Wen’s Foodstuffs Co., Ltd., for phenotype measurements and other contributions to our study.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript. Yang M, Cai G, Wu Z are employees of Guangdong Wens Foodstuffs Co., Ltd..