Hemicastration induced spermatogenesis-related DNA methylation and gene expression changes in mice testis
Article information

Abstract
Objective
Hemicastration is a unilateral orchiectomy to remove an injured testis, which can induce hormonal changes and compensatory hypertrophy of the remaining testis, and may influence spermatogenesis. However, the underlying molecular mechanisms are poorly understood. Here, we investigated the impact of hemicastration on remaining testicular function.
Methods
Prepubertal mice (age 24 days) were hemicastrated, and their growth was monitored until they reached physical maturity (age 72 days). Subsequently, we determined testis DNA methylation patterns using reduced representation bisulfite sequencing of normal and hemicastrated mice. Moreover, we profiled the testicular gene expression patterns by RNA sequencing (RNA-seq) to examine whether methylation changes affected gene expression in hemicastrated mice.
Results
Hemicastration did not significantly affect growth or testosterone (p>0.05) compared with control. The genome-wide DNA methylation pattern of remaining testis suggested that substantial genes harbored differentially methylated regions (1,139) in gene bodies, which were enriched in process of protein binding and cell adhesion. Moreover, RNA-seq results indicated that 46 differentially expressed genes (DEGs) involved in meiotic cell cycle, synaptonemal complex assembly and spermatogenesis were upregulated in the hemicastration group, while 197 DEGs were downregulated, which were related to arachidonic acid metabolism. Integrative analysis revealed that proteasome 26S subunit ATPase 3 interacting protein gene, which encodes a protein crucial for homologous recombination in spermatocytes, exhibited promoter hypomethylation and higher expression level in hemicastrated mice.
Conclusion
Global profiling of DNA methylation and gene expression demonstrated that hemicastration-induced compensatory response maintained normal growth and testicular morphological structure in mice.
INTRODUCTION
The testes play an important role in reproduction, which are responsible for the generation of male gametes and the production and release of sex steroids. Hemicastration is a unilateral orchiectomy to remove a testis injured by external force, biochemistry or illness. Hemicastration of prepubertal mammals induces hypertrophy of the remaining testis and may be associated with changes in serum follicle-stimulating hormone levels [1]. Furthermore, researches have revealed that hypertrophy of the remaining testis results from rapid proliferation of Leydig and Sertoli cells [2]. These effects on testicular function have also been demonstrated: unilateral orchiectomy restored spermatogenesis in nonrelapsing testicular cancer cases after 1 year, even in patients exhibiting oligospermia or azoospermia [3]. It has also been reported that hemicastration improved the quality and volume of semen in male Iraqi goats [4]. However, hemicastration of sexually vigorous rats reduced sexual behavior and sperm count [5]. Hemicastration has long been used as an important way for testicular development research, and such knowledge would also help to improve reproductive efficiency in livestock. Nevertheless, the mechanism of compensatory response and the consequent influence on spermatogenesis in the remaining testis is still poorly understood.
Epigenetic modifications mainly include DNA methylation, histone modification and nucleosome positioning. DNA methylation is essential for genome-wide pre-transcriptional regulation, which is involved in numerous biological processes. A previous study reported that castration induced demethylation, primarily in tissue-specific enhancer sequences, exclusively in the male liver [6]. Furthermore, castration also alters the DNA promoter methylation status of some steroid-responsive genes in the rat brain, and may influence homeostatic regulation of behavioral relevant systems [7]. Moreover, giving neonatal mice testosterone drives abundant sexually dimorphic cytosine–phosphate–guanine (CpG) sites to masculinize in brain [8]. Nevertheless, whether hemicastration affects steroid hormone biosynthesis or induces DNA methylation changes in the remaining testis is unknown.
In this study, we performed unilateral orchiectomy on Kun ming (KM) mice at weaning. When the mice reached physical maturity, we used high-throughput reduced representation bisulfite sequencing (RRBS) and RNA sequencing (RNA-seq) to explore the DNA methylation and expression changes in the remaining testis. We found that hemicastration caused differential expression of a number of genes involved in spermatogenesis and prostaglandin biosynthesis, and that these genes were partially regulated by DNA methylation.
MATERIALS AND METHODS
Animal ethics statement
All procedures involving animals in this study were conducted following the rules of the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in June 2004) and approved by the Institutional Animal Care and Use Committee of the College of Animal Science and Technology, Sichuan Agricultural University, Sichuan, China under permit No. DKY-S20153352.
Animals and sample collection
Male KM mice were randomly divided into control (n = 19) and hemicastration (n = 19) groups after weaning (age 24 days), and were housed and maintained on a 12 h:12 h light:dark cycle, with free access to standard chow and water. KM mice originated from the Swiss mice, which has been wildly used for experiment in China, such as the model for studying age-related decline in fertility [9]. The mice were anesthetized with ethyl ether and unilateral orchiectomy was performed via a midline scrotal incision to allow unilateral (left) testis removal in the hemicastration group [10]. At the same time, sham surgery was performed on the control group. The incisions were sutured then disinfected with potassium permanganate powder. When the mice reached age 72 days (physical maturity), final measurements were taken and the animals were sacrificed. The mice volume was measured by drainage method. The right testis was rapidly separated from each carcass and immediately frozen in liquid nitrogen, and stored at −80°C until DNA or RNA extraction.
Serum measurements
Blood samples were collected via eye enucleation, and were left to stand for 1h at 4°C. Subsequently, the collected blood samples were centrifuged at 3,000×g at 4°C for 30 min to separate serum for further analysis. Serum testosterone concentration was measured using a commercial radioimmunoassay kit (Beijing North Institute of Biological Technology, Beijing, China).
Testis histology
For histological analysis, the testicular tissues with 10% paraformaldehyde treatment were embedded in paraffin, and sectioned at 5 μm thicknesses, then stained with Hematoxylin and Eosin. Representative photomicrographs were taken with a light microscope.
RNA and DNA isolation, library preparation and sequencing
Because of the high consistency among individuals within each group, we randomly chose 3 biological replicates out of each group to represent the control and hemicastration for RRBS, of which two were used for RNA-seq. DNA of testis was extracted using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. DNA with more than 5 μg initial volume and an ratios of optical density at 260 to that at 280 nm (OD260/280) value between 1.8 and 2 for each sample was regarded as suitable for subsequent RRBS experiments. We constructed mice RRBS libraries according to previously published protocols [11] with slightly modification. Briefly, genomic DNA was digested by the restriction enzyme MspI. Subsequently, the Illumina paired-end protocol, which comprises end-repair, A-tail addition and methylated adaptor ligation, was performed to construct libraries. Fragments (40 to 220 bp) were selected and purified, then treated with bisulfite using a Zymo EZ DNA Methylation Gold kit (Zymo Research, Orange, CA, USA). All bisulfite-treated products were amplified by polymerase chain reaction (PCR) for library preparation, then resulting library was sequenced with Illumina HiSeq4000.
Total RNA was extracted from mice testis using Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Agarose gel electrophoresis and Agilent 2100 (Agilent Technologies, Foster City, CA, USA) were used to assess RNA integrity, and Nanodrop (Implen, Westlake Village, CA, USA) and Qubit (Life Technologies, Foster City, CA, USA) were used to measure concentration. Subsequently, RNA from each animal was used for RNA-seq library construction through NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, Ispawich, MA, USA) following manufacturer’s instructions. Briefly, poly(A) RNA enrichment of total RNA was performed using oligo (dT) magnetic beads, then the resultant fragments were used as the template for reverse transcription with random hexamer primers to synthesize first-strand cDNA. This was then supplemented with buffer, dNTPs, DNA polymerase I and RNase H to produce second-strand cDNA. The double-stranded cDNA was purified using AMPure XP beads (Beckman Coulter, Beverly, MA, USA), then subjected to end-repair, A-tail addition, ligation to paired-end Illumina sequencing adaptors, fragment size selection by AMPure XP beads and amplification by PCR for library preparation. The high-quality library preparations were sequenced using an Illumina HiSeq platform and paired-end reads were generated.
Read mapping and data processing
Filtered data were obtained by removing reads containing adaptor, poly-N sequences, or low quality reads, from the raw data. The Q20, Q30 and GC content of the clean data were calculated. Subsequently, the high-quality data were mapped to the mouse reference genome (Mus_musculus, GRCm38.81) using Tophat (version 2.1.0) and Bismark (version 0.12.5) for RNA-seq and RRBS, respectively. When multiple reads from one sequencing library were mapped to same genomic location, these reads were regarded as potential clonal duplicates arising from PCR amplification biases and were considered a single read.
For RRBS, we defined the region extending from −2,200 to +500 bp from the gene transcription start site (TSS) as the promoter. Meanwhile, gene body regions was defined as +1 kb from the TSS to −1 kb from the transcription termination site (TTS). We calculated methylation level of methylated cytosine across the chromosomes with sliding-window approach (window size 3,000 bp and step size 600 bp). Methylation level (ML) = (number of methylated cytosines within the sliding window)/(number of cytosines within the sliding window regardless of methylation status). For RNA-seq, Cufflinks (version 2.2.1) was used to quantify gene expression and obtain fragments per kilobase of transcript per million mapped reads expression values.
The RNA-seq and RRBS data have been deposited into the NCBI gene expression omnibus under the accession GSE95694.
Identification of differentially methylated region and differentially expressed genes
Differentially methylated region (DMRs) were identified using BSseq (part of the R package), following the criteria that the region should contain at least three CpG sites and variation between the two groups should be more than 10%. DEseq2 (part of the R package) was used to detect genes in the testis that were differentially expressed between the control and hemicastration groups. Genes with Benjamini-Hochberg adjusted p-values <0.05 were deemed as differentially expressed genes (DEGs).
Gene functional enrichment analyses
Genes containing DMRs and DEGs and were uploaded to the DAVID webserver (https://david.ncifcrf.gov/) for functional enrichment analysis for gene ontology (GO) terms involving molecular function, biological processes and the Kyoto encyclopedia of genes and genomes (KEGG) pathway (Benjamin-corrected p-value <0.05).
Quantitative polymerase chain reaction analyses
Total RNA was extracted from the testis that corresponded to the sample used for sequencing using Trizol reagent, and cDNA was synthesized using a PrimeScript 1st Strand cDNA Synthesis kit (Takara Bio, Otsu, Shiga, Japan), following the manufacturer’s protocol. Subsequently, the cDNA sample was analyzed in triplicate by quantitative PCR (qPCR) performed on a CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA, USA) using SsoFast EvaGreen Supermix (Bio-Rad, USA) according to the manufacturer’s protocol. The reaction mixture for qPCR (10 μL) comprised 5 μL SsoFast EvaGreen Supermix, 0.5 μL each of sense and antisense primer, 1 μL cDNA and 3 μL RNase-free water. The following PCR conditions were used for all genes: 95°C for 3 min, followed by 40 cycles of 95°C for 10 s, optimal amplification temperature for 30 s, then 95°C for 30 s. A melting curve (65°C to 95°C) was established to test the specificity of genes. The geometrical mean of expression values for three endogenous control genes (ekaryotic translation elongation factor 2 [EEF2], peptidylprolyl isomerase A [PPIA], and β-ACTIN) was used to correct the gene expression data. The 2−ΔΔCT method was used to analyze the qPCR data and calculate relative expression. The primers used for measuring the expression of candidate mRNAs are listed (Supplementary Table S1).
RESULTS AND DISCUSSION
Phenotypic measurements
To investigate the effect of hemicastration on mouse growth and development, we evaluated body weight, growth parameters, right testis ratio (right testis weight/body weight with both testes removed) and serum testosterone levels and found no significant differences (p>0.05) between hemicastrated and control mice (Table 1). Comparative histological analysis of testis section didn’t display obvious aberrant tubular architecture and size in hemicastrated and control mice, as did germ cells were not lacking (Supplementary Figure S1). Testosterone regulates multiple biological processes, and its deficiency induced by castration would influence multi-index related to growth of animals such as body weight and testicular development [12,13]. In present study, hemicastration did not reduce the concentration of serum testosterone, as well as testicular histology and other measurements, which is consistent with previous study [14]. These observations indicate that hemicastrated mice maintain normal body growth and testicular morphological structure despite unilateral absence of testis. However, it takes further work to study whether hemicastration affects mice fertility, which requires semen analysis and offspring evaluation.
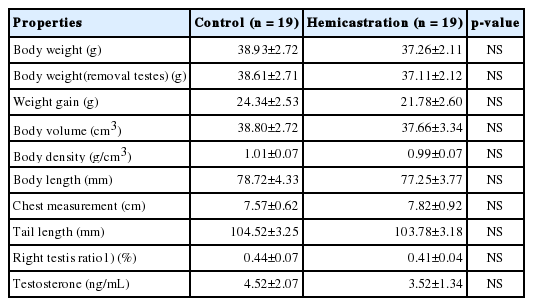
Phenotypic characteristics of experimental mice
Hemicastration induced different DNA methylation in testis
DNA methylation is an important epigenetic modification to regulate genes expression to response external stimulation. It has been reported that most methylation changes happened in genes promoter regions are negatively correlated with genes expression, while the changes happened in genes body regions may have opposite function [15]. To investigate DNA methylation changes in the testis in hemicastrated mice, we performed RRBS on a total of six samples (three replicates for each group). The mapped rate of each library was more than 80% (Supplementary Table S2). The hierarchical clustering of DMRs distinguished the two groups (Figure 1A), and correlated rates of biological replicates (average Pearson’s r = 0.95) were higher than treatment (average Pearson’s r = 0.93) (Supplementary Figure S2). More DMRs were enriched in gene bodies (1,139 DMRs exist in 1,249 genes) than in promoters (237 DMRs exist in 243 genes) (Figure 1B). These results may indicate relative epigenetic concordance within each group and noticeable hemicastration induced DNA methylation changes.
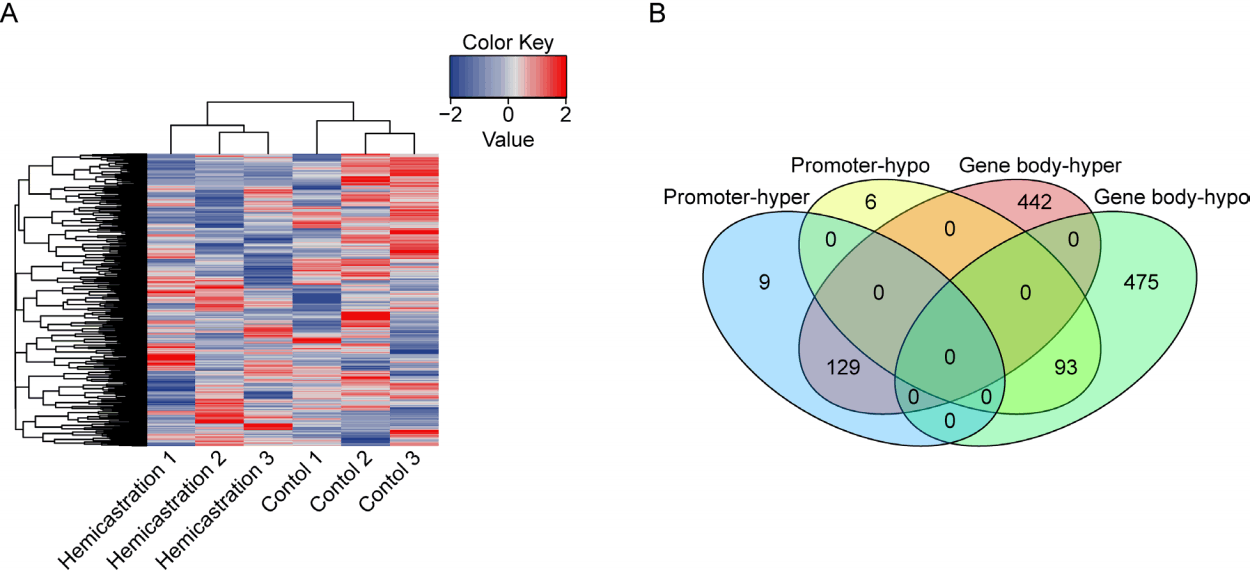
Hierarchical clustering and distribution of differentially methylated regions (DMRs). (A) Hierarchical clustering of DMRs in individuals. Red, DMR with hypermethylation; blue, DMR with hypomethylation. (B) Venn diagram of locations of DMRs that were hyper- and hypomethylated after hemicastration.
Genes with DMRs involved in spermatogenesis
We performed enrichment analysis to investigate the potential function of genes with different methylated regions (Figure 2). Genes with DMRs in their gene bodies were significantly enriched in GO categories and pathways related to ‘protein binding’ (n = 281, p = 1.75×10−8), ‘cell adhesion’ (n = 46, p = 8.38×10−5) and ‘regulation of actin cytoskeleton’ (n = 24, p = 3.4×10−4). Assembly and disassembly of Sertoli–Sertoli and Sertoli–germ cell junction involves protein binding, cell adhesion and regulation of actin cytoskeleton, which plays an indispensable role during spermatogenesis [16]. Cell junction between Sertoli cells produces the blood–testis barrier, and relates to tethering of the proteins claudin and occludin to the actin cytoskeleton by adaptor proteins such as zonula occludens-1. It has been reported that knocking out the genes that encode these proteins diminishes tight junction formation and results in increased apoptosis and seminiferous tubule atrophy, leading to massive loss of germ cells and infertility in mature mice [17]. Additionally, disordered Sertoli–germ cell adhesion disrupts germ cell localization and causes premature release of spermatids into the lumen, thus resulting in an abnormal elongation process and infertility [18]. Our results indicate that, to some degree, hemicastration induced genes methylation changes in remaining testis which may participate in spermatogenesis process.
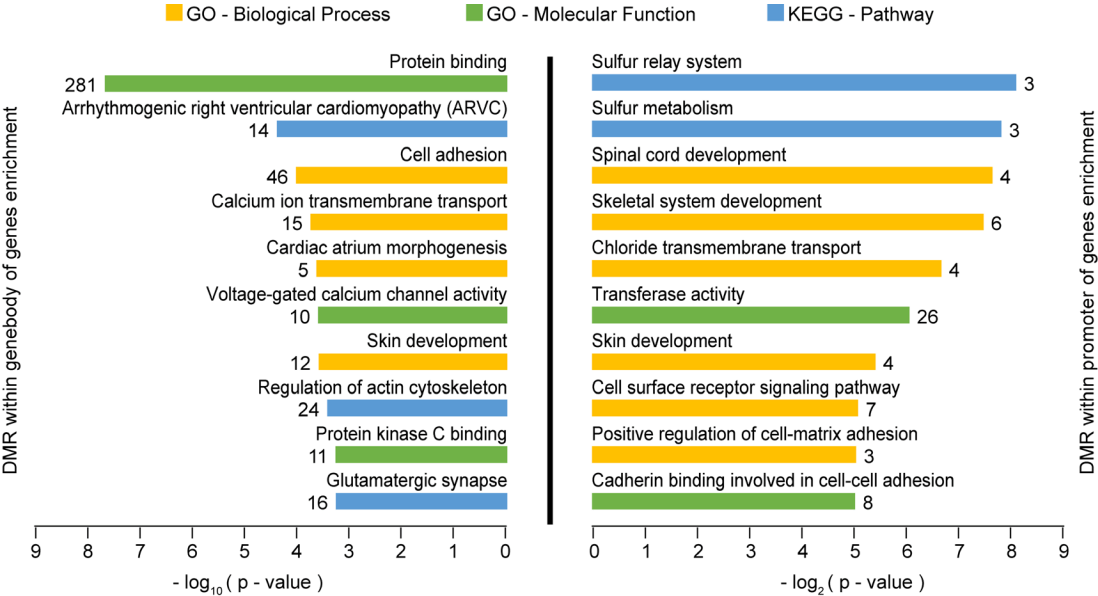
Functional enrichment analysis of differentially methylated regions (DMRs). Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis of genes harboring DMRs. The top 10 terms of genes with p-value<0.05 are listed. The number next to each bar represents the number of genes.
Testicular transcriptome analysis
For exploration of genes expression induced by DNA methylation and further insight into the molecular mechanisms underlying the influence of hemicastration on spermatogenesis, we performed RNA-seq on two testicular samples from each group (hemicastrated and control) and generated approximately 5 giga bases (Gb) of filtered data for each sample. For each sample, approximately 89% of the total reads could be mapped to the mouse reference genome (Supplementary Table S3). Meanwhile, correlation rates of biological replicates (average Pearson’s r = 0.99) was greater than treatment (average Pearson’s r = 0.98) (Supplementary Figure S2), which suggested experimental reliability.
We detected 243 DEGs after hemicastration under the criterion p<0.05 by DEseq2, 46 of which were upregulated, while 197 were downregulated in the hemicastration group (Figure 3A). We then performed qPCR to validate RNA-seq results (Figure 3B), and implemented functional enrichment analyses by querying the DEGs that were upregulated and downregulated by hemicastration to gain further insight into their biological functions (Figure 3C). The most represented GO categories for upregulated DEGs were ‘meiotic cell cycle’ (n = 5, p = 2.5×10−3), ‘synaptonemal complex assembly’ (n = 3, p = 2.85×10−2) and ‘spermatogenesis’ (n = 6, p = 3.39×10−2). Synaptonemal complex assembly is the core process of the meiotic cell cycle because of its key roles in the pairing, synapsis and segregation of chromosomes and the exchange of the axial elements of the two chromatids in pachytene spermatocytes. It has been reported that synaptonemal complex protein 1 (SYCP1), synaptonemal complex protein 3 (SYCP3), and HORMA domain containing 1 (HORMAD1) are required for synaptonemal complex formation [19]. Mutation of any one of these three genes disrupted homologous chromosome pairing and increased spermatocyte apoptosis, thereby resulting in early meiotic arrest of spermatogenesis [20]. After meiosis, round spermatids undergo spermiogenesis to transform into elongated spermatids. Meiosis-expressed gene 1 (MEIG1) plays an important role in spermiogenesis, which forms a complex with other proteins and binds to manchette, then transports cargos along the manchette to build sperm flagella [21]. MEIG1-deficency disrupted the manchette and substantially reduced elongated spermatid number [22]. SYCP1, SYCP3, HORMAD1, and MEIG1 showed coordinated upregulation of expression levels in the hemicastrated mouse (Supplementary Figure S3), which indicates a normal spermatogenesis process in remaining testis after hemicastration.
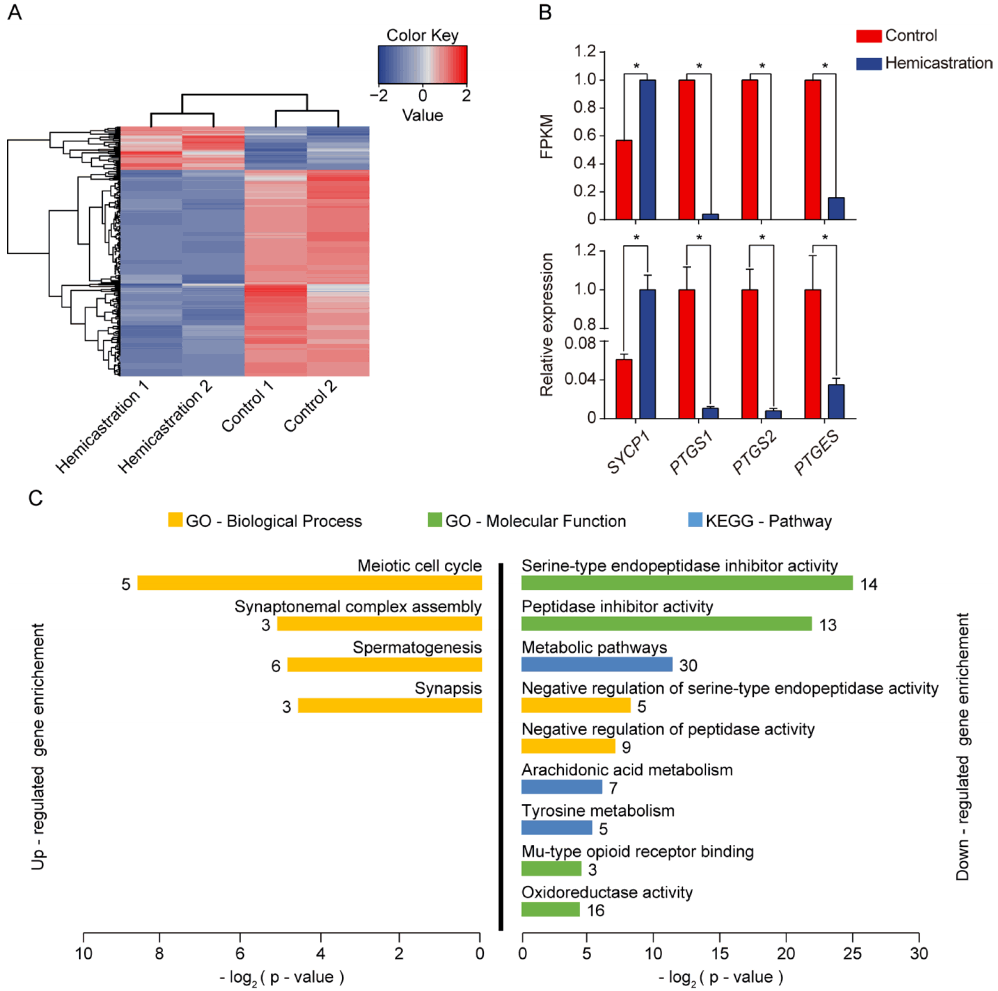
Testicular transcriptomic analysis. (A) Hierarchical clustering of differentially expressed genes (DEGs) in individuals. Red, upregulation gene; blue, downregulation gene. (B) Quantitative polymerase chain reaction (qPCR) of four candidate genes. The value of expression is normalized. (* p<0.05). (C) Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis of DEGs that were up- and downregulated after hemicastration. Terms with Benjamini-corrected p-values<0.05 are listed. The number next to each bar represents the number of DEGs.
Partial downregulated DEGs were enriched in the GO cate gories ‘serine-type endopeptidase inhibitor activity’ (n = 14, p = 3.21×10−8) and ‘negative regulation of serine-type endopeptidase activity’ (n = 5, p = 3.42×10−3). Sertoli cells are responsible for secretion of proteases and protease inhibitors to facilitate germ cell migration [23]. The dynamic inverse relationship between proteases and protease inhibitors is indispensable for Sertoli–germ cell junction disassembly to release elongated spermatids into the tubule lumen [24]. Coincidentally, in the present enrichment analysis, we found that hemicastration increased expression of genes relevant to endopeptidase activity but reduced those related to endopeptidase inhibition, which suggests that hemicastration may accelerate the release of elongated spermatids in the remaining testis. Meanwhile, enrichment analysis also indicated that the some downregulated DEGs were enriched in the KEGG pathway ‘arachidonic acid metabolism’ (n = 7, p = 1.49×10−2). Prostaglandin-endoperoxide synthase 1 (PTGS1), prostaglandin-endoperoxide synthase 2 (PTGS2), and prostaglandin E synthase (PTGES) play important roles in the process of arachidonic acid convert to prostaglandin, which consumes phospholipid of cell membrane [25]. In other words, reduction of prostaglandin may mean the accumulation of phosphocholine. A previous study reported that fertile men have higher concentrations of phosphocholine than patients with Sertoli cells only, which suggests that the production of sperm requires high levels of phospholipid to support constant membrane synthesis [26]. Our RNA-seq and qPCR results both indicate that expression of PTGS1, PTGS2, and PTGES declined after hemicastration (Figure 3B), which may imply the enhanced spermatogenesis with increased membrane synthesis.
In general, the changes observed in the global gene expression pattern may suggest that hemicastration could reinforce gene expression, which related to promote synapsis during meiosis and spermiogenesis and adjust relevant metabolic homeostasis in the testis. These evidences may demonstrate normal testicular function of hemicastrated mice.
Differentially expressed genes that harbor DMRs
We analyzed the DNA methylation profile in conjunction with the expression pattern to identify genes that were regulated by DNA methylation changes. However, only 11 of the DEGs harbored DMRs (Supplementary Figure S4). DNA methylation serves as a pre-transcriptional epigenetic modification, and may not exclusively determine the expression of relevant genes because other regulation mechanisms exist. Unexpectedly, we found a DMR (109 bp width and containing 9 CpGs) located in the proteasome 26S subunit ATPase 3 interacting protein (PSMC3IP) promoter, which was hypomethylated after hemicastration, and exhibited an increased expression level (Figure 4). This finding is agreement with the previous reported that hypomethylation in promoter region may promote genes transcription. PSMC3IP encodes the protein Hop2, a subunit of the Hop2–Mnd1 complex, which plays an important role in homologous recombination [27]. It has been reported that Hop2–Mnd1 serves as a cofactor in strand exchange, and ensures synaptic alignment and D-loop formation [28]. Knocking out Hop2 (PSMC3IP) induced spermatocyte arrest at the pachytene stage of meiotic prophase I, inhibited repair of meiotic double-strand breaks, and resulted in male infertility [29].
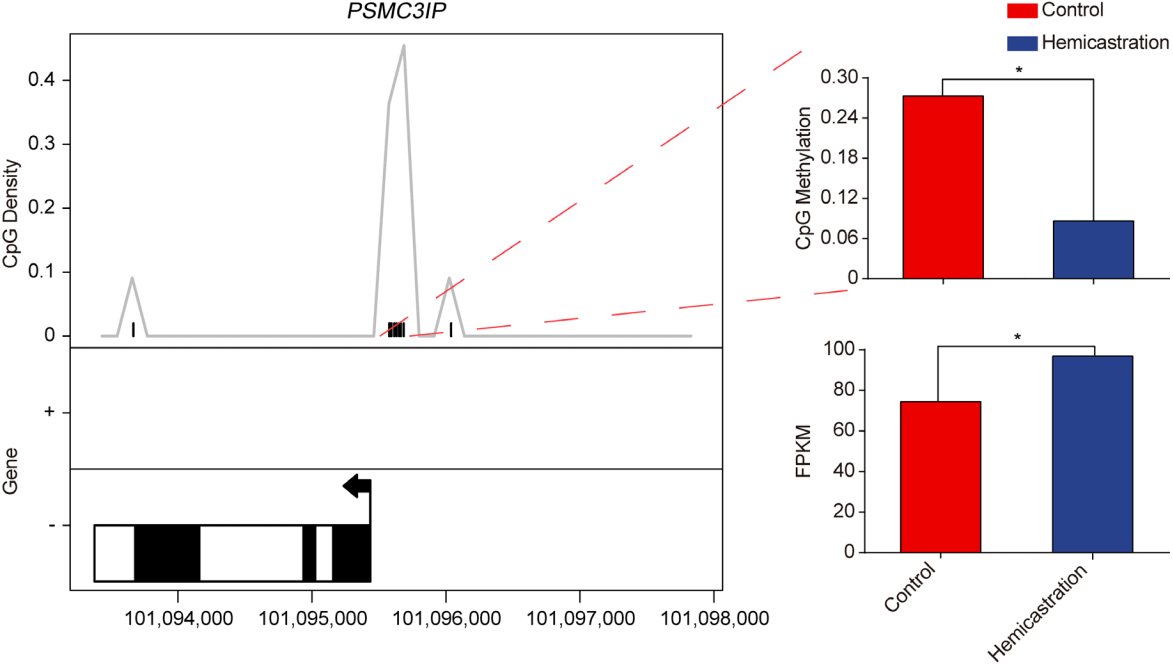
Differentially methylated region (DMR) and gene expression of proteasome 26S subunit ATPase 3 interacting protein (PSMC3IP). Left, locations of DMR and PSMC3IP; solid box indicates exon; hollow box indicates intron. Upper right: cytosine–phosphate–guanine (CpG) methylation of DMR. Lower right: fragments per kilobase of transcript per million mapped reads (FPKM) of PSMC3IP.
Epigenetic modification plays an important role in gametogene sis, it has been reported the absence of DNA methyltransferase 3-like (Dnmt3L) caused overall loss of cytosine–guanine (CG) and non-CG methylation in germ cells and resulted in infertility. Meanwhile, ten eleven translocation 1 (TET1) 5-methylcytosine dioxygenase mutant increased promoter methylation level of meiosis-related genes and reduced corresponding genes expression [30]. Besides, histone H3 lysine 9 (H3K9) methyltransferase G9a regulated spermatogenesis through control methylation of H3K9. However, which metabolites were induced by hemicastration and how them regulate corresponding genes expression and influence spermatogenesis needs to be further studied.
In the present study, promoter hypomethylation and the up regulation of the PSMC3IP may indicate that hemicastration enhanced homologous recombination during meiosis and promoted germ cell differentiation.
CONCLUSION
In summary, we demonstrated that hemicastrated mice still maintained normal growth and testicular morphological structure. The global DNA methylation changes observed may also indicate the role of this epigenetic modification in regulating genes after hemicastration. Moreover, transcriptome analysis indicated that expression of meiosis-related genes did not decrease after hemicastration, and even tended to increase, which suggests that a compensatory response may enhance testicular function in mice in the absence of one testis. However, whether hemicastrated mice maintained normal fertility and biological mechanism underlying this compensation remains to be determined. Our observations provide a molecular basis for understanding the influence of hemicastration on testicular function.
Supplementary Information
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (31601919, 31522055 and 31530073), the Program for Innovative Research Team of Sichuan Province (2015TD0012), Sichuan Province & Chinese Academy of Science of Science & Technology Cooperation Project (2017JZ0025), the Project of Sichuan Education Department (15ZA0003 and 16ZB0037), the National Program for Support of Top-notch Young Professionals, the Young Scholars of the Yangtze River.
Notes
AUTHOR CONTRIBUTIONS
Yixin Wang, Jideng Ma, Mingzhou Li, and Long Jin conceived and designed the experiments. Yixin Wang, Li Chen, and Keren Long performed the experiments. Yuhua Fu, Yixin Wang, Long Jin, Silu Hu, Yang Song, Dazhi Shang, and Qianzi Tang performed the bioinformatics analysis process. Mingzhou Li, Xuewei Li, and Xun Wang contributed reagents/materials/analysis tool. Yixin Wang and Long Jin wrote the paper. Mingzhou Li revised the paper. All authors reviewed the manuscript.
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.