




INTRODUCTION
Yak (Bos grunniens) is the only bovine species adapted to the special high-altitude ecological environment of the Qinghai-Tibet Plateau, and semi-domestic yaks provide the basic resources such as meat, milk, transportation, dung for fuel and hides for tented accommodation that are necessary for Tibetans and other nomadic pastoralists living there [1]. Herbivorous mammals rely on resident gut microorganisms to gain energy from their main food sources, and this has entailed major changes in digestive anatomy and physiology that allow efficient microbial fermentation to take place alongside the recovery of dietary energy by hosts. Ruminants (foregut fermentors) benefit from microbial protein as well as the absorption of energy that is released by anaerobic microorganisms in the form of fermentation acids [2]. The semi-open farming system and the lack of sufficient feed stuff in rangelands during winter and spring seasons lead to yaks’ slow growth due to insufficient nutrients, which may result in a series of problems, such as weight losing, sickness, and even death. While there is the potential to resolve these problems by feeding supplementary silage forage with microbial agents to regulate the gastrointestinal microbiota of yaks, which may promote the normal growth of yaks. Consequently, it is crucial to understand the important relationship of gut microbiota with the healthy growth and disease control in yaks. However, few studies have reported this [3].
The microbial flora in animals’ gastrointestinal tract is a unique and diverse ecosystem, which has one of the highest cell density systems found so far [4]. The microbial diversity and functional redundancy of gut microbial flora may contribute to the stability of this dynamic ecosystem. Non-pathogenic gut microbiota can protect the intestine from infection of harmful microbes, while the symbiotic ones can promote the health of hosts and thus to improve the productivity of animals [5]. The metabolic potential of gut microbiota in ruminants produces energy from the extraction of indigestible carbohydrates (dietary fiber) and conversion of the host-derived substances, non-nutritive dietary components. Gut microbiota shares specialized relationships with their hosts, and to a certain extent, genomics can reveal the dynamics of these correlations [6].
Denaturing gradient gel electrophoresis (DGGE) fingerprint technique has a unique superiority in the study of genetic diversity and population difference of complex microbiota, and it has been widely applied [7,8]. Metagenomic sequencing represents a powerful alternative to ribosomal RNA (rRNA) sequencing for analyzing complex microbial communities [9], and this method avoids the need to culture individual microorganisms and eliminates cloning and polymerase chain reaction (PCR) biases [10]. Recently, Patel et al [11] found that diet proportion, fraction of rumen, and forage type affected rumen microbiome of Indian Kankrej cattle at taxonomic as well as functional levels by metagenomic sequencing. According to metagenomic analysis results, Zhu et al [6] found that ~37-Mbp contig sequences from gut microbes of the giant panda recovered putative genes coding cellulose-digesting related enzymes in Clostridium group I, showing that the giant pandas had evolved a number of traits to overcome the anatomical and physiological challenge of digesting a diet high in fibrous matter.
Thus, the objective of present study was to reveal the dynamic changes of the gut microbial community during yaks’ growth and to understand the benefits of gut microbiota on nutrient metabolism of hosts by PCR-DGGE and metagenomic sequencing, in order to assist the searching for potential probiotics which could be beneficial to yaks’ growth through supplementary feeding with these probiotics agents.
MATERIALS AND METHODS
Sample collection
Gut content samples from 0.5, 1.5, and 2.5 years old of male yaks were collected immediately after slaughtering, snap-frozen in liquid N2, and shipped to the laboratory. All samples were obtained inside the colon, where there was no contact with infectious microbes. Five gut content samples from each age of semi-domestic yaks were collected from the local abattoir of Hongyuan County in Aba Tibetan and Qiang Autonomous Prefecture, Sichuan province. Based on the research guidelines, these yaks were managed as one group in a large lot owned by Sichuan Grassland Science Academy.
DNA extraction and polymerase chain reaction amplification
DNA was extracted from 15 gut contents samples (5 yaks in each age group) using the Stool DNA Kit (OMEGA, Norcross, GA, USA) according to the protocol for isolation of DNA for pathogen detection. DNA was eluted in a final volume of 200 μL using elution buffer and then stored at −20°C. Tubes containing only the Stool DNA Kit extractions were included throughout the lysis and PCR steps to serve as negative controls. For PCR-DGGE analysis of the total bacteria, Nested PCR was used to amplify the V2–V3 region of the 16S rRNA gene. In the first PCR amplification, the universal bacterial 27F and 1492R primers were used to amplify the 16S rRNA gene in a S1000 thermal cycler (Bio-Rad, Hercules, CA, USA) using the following program: initial denaturation for 5 min at 94°C; 30 cycles of 94°C for 30 s, 58°C for 45 s, and 72°C for 90 s; final elongation for 7 min at 72°C. The PCR reaction solution (50 μL total) contained 2 μL of DNA template (50 ng/μL), 2 μL of each primer (10 μM), 19 μL of ddH2O and 25 μL of 2×Taq PCR MasterMix (contained 500 μM dNTP, 0.1 U Taq polymerase/μL, 20 mM Tris-HCl, 100 mM KCl and 3 mM MgCl2) (TIANGEN, Beijing, China). Then 16S rDNA was purified using the Gel Extraction Kit (OMEGA, USA) according to the standard protocol. In the second PCR amplification, the universal bacterial HDA1-GC and HDA-2 primers [12] were used to amplify the V2–V3 region of the 16S rRNA gene in a S1000 thermal cycler (Bio-Rad, USA) using the following program: initial denaturation for 5 min at 94°C; 30 cycles of 94°C for 30 s, 53°C for 30 s, and 68°C for 30 s; final elongation for 7 min at 68°C. The PCR reaction solution (50 μL total) contained 2 μL of DNA template (50 ng/μL), 2 μL of each primer (10 μM), 19 μL of ddH2O and 25 μL of 2×Taq PCR MasterMix (contained 500 μM dNTP, 0.1 U Taq polymerase/μL, 20 mM Tris-HCl, 100 mM KCl and 3 mM MgCl2) (TIANGEN, China). The sequence of the GC clamp was 5′-CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGG-3′ [13].
Denaturing gradient gel electrophoresis analysis
The PCR products (50 μL each line) were subjected to DGGE analysis using a 35% to 65% gradient with a 8% acrylamide gel run at 120 V, 60°C for 6 h with the DCode Universal Mutation Detection System (Bio-Rad, USA). After electrophoresis, the gel was stained with SYBR Green I (1:10,000 dilution in tris-acetic acid ethylenediaminetetraacetic acid buffer, Sigma, Billerica, MA, USA) in darkness for 45 min (three times, 15 min each), viewed with a Gel Imaging System (Universal Hood II, Bio-Rad, USA), and photographed. The similarities and differences in the microbial structure were determined by comparing the clusters of the whole DGGE profiles using the Quantity One software package (Version 4.6.9, Bio-Rad, USA). The similarity matrices were produced using the Dice similarity coefficient, which allowed for the construction of dendrograms using the unweighted pairwise grouping method with mathematical averages (UPGMA) method [14]. Principal component analysis (PCA) was performed (on mean-centered data) to visualize the general structure of species-level composition of gut microbiome using the Canoco for Windows 4.5 Software.
Metagenomics analysis
DNA was extracted from gut content of 15 yaks (5 yaks in each age group) using the Stool DNA Kit (OMEGA, USA) according to the standardized protocol. Sequencing and general data analyses were performed by Macrogen (Seoul, Korea). DNA library construction and sequencing followed BGI’s previous work on human gut microbe metagenomic sequencing using a HiSeq 2000 system (Illumina, San Diego, CA, USA) [9]. We compared the raw short reads with yak genome data to remove the host sequence. The clean reads thus obtained were assembled to obtain the long contig sequences by the short oligonucleotide analysis package (SOAP) de novo assembler [15] as used in human gut microbe metagenomic analyses. We tried different K-mer frequencies to obtain different assembly results, and used N50 lengths to access the best assembly results.
Gene prediction and taxonomic assignment
We used the assembly contig sequences and applied MetaGene software, with only open reading frame (ORF) longer than 100 bp preserved. The ORFs were translated into protein sequences using National Center for Biotechnology Information (NCBI) Genetic Code 11. We carried out BLASTP [16] alignment to query the predicted protein sequences against the integrated NR protein database. For each predicted gene, hits with E-values >1×10−5 were filtered. Then a significant-matches set was retained to distinguish taxonomic groups, which were defined for hits with E-values <10 times the top hit E-value. Next, the lowest common ancestor (LCA)-based algorithm implemented in MEGAN [17] was introduced to determine the taxonomic level of each gene. The LCA-based algorithm assigned genes to taxa so that the taxonomic level of the assigned taxon reflected the level of conservation of the gene.
Gene functional classification
We performed predicted gene functional classification by querying protein sequences of the genes against the Kyoto encyclopedia of genes and genomes (KEGG) database using BLASTP with E-values <1×10−5. Genes were annotated as a function of the KEGG homologs with the lowest E-value. In the KEGG database, genes were assigned to KEGG pathways.
Statistical analysis
The statistical analysis of metagenomic profiles (STAMP; version 2.0.2) statistical probability model was employed to identify biologically relevant differences between metagenomic communities [18]. This model allows choosing appropriate statistical methods to evaluate differences in the proportions of sequences assigned to different taxonomic groups between metagenomes, while considering effect sizes and confidence intervals in assessing biologically relevant differences. Two-way comparisons of taxonomic distributions between metagenomic samples were tested within STAMP, using the Fisher’s exact test associated with the Newcombe-Wilson method for calculating confidence intervals (nominal coverage of 95%). Corrected p-values were calculated using the Bonferroni correction.
RESULTS
Comparison of detectable bacteria in the gut from different age of yaks using PCR-DGGE analysis
To identify detectable species of microbes present in the gut from different age of yaks, we applied PCR-DGGE profiles analysis. Nested PCR was used to amplify the V2–V3 region of the 16S rRNA gene. More than 40 bands were resolved, and unique bands in the gel were identified by sequencing and annotated from NCBI BLAST database (data not shown) (Figure 1A). According to the DGGE fingerprint, the similarity matrices were produced using the Dice similarity coefficient and the dendrogram was constructed using UPGMA clustering algorithm (Figure 1B). On the other hand, bacterial communities were clustered using PCA, which distinguished microbial communities based age of gut sampling. As shown in Figure 1C, PCA disclosed that animal’s age promoted the main change in gut microbiota of yaks. Gut metagenomes from the same age group clustered very distinctly from those of different ages, indicating that the age had a substantial effect on the microbial composition in yak gut.
Phylogenetic compositions of the gut bacterial communities in yaks
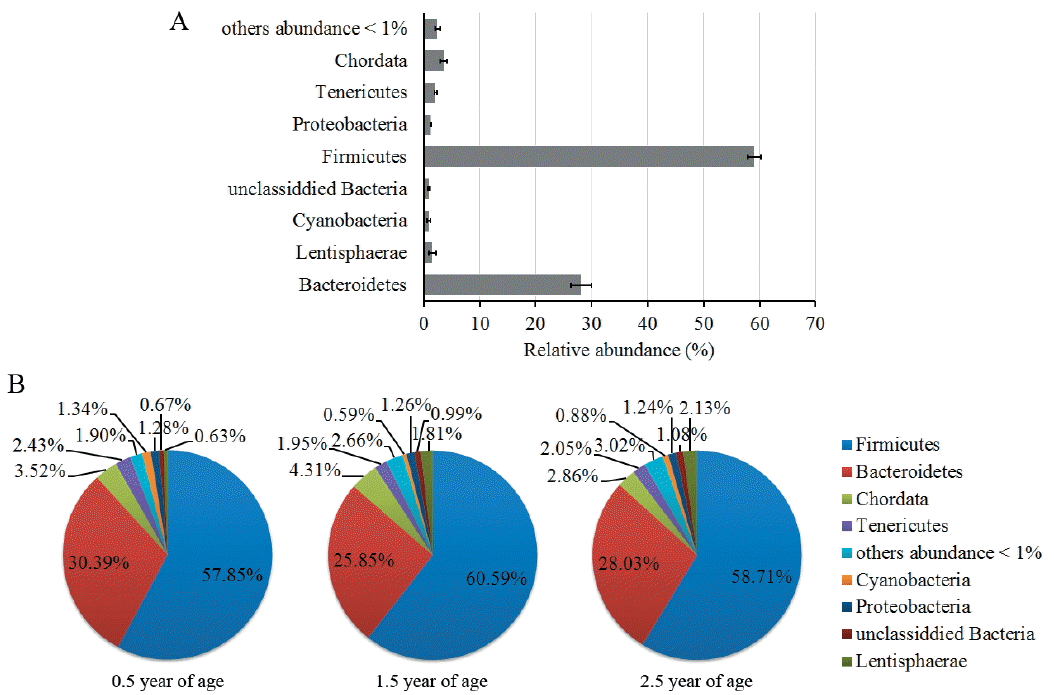
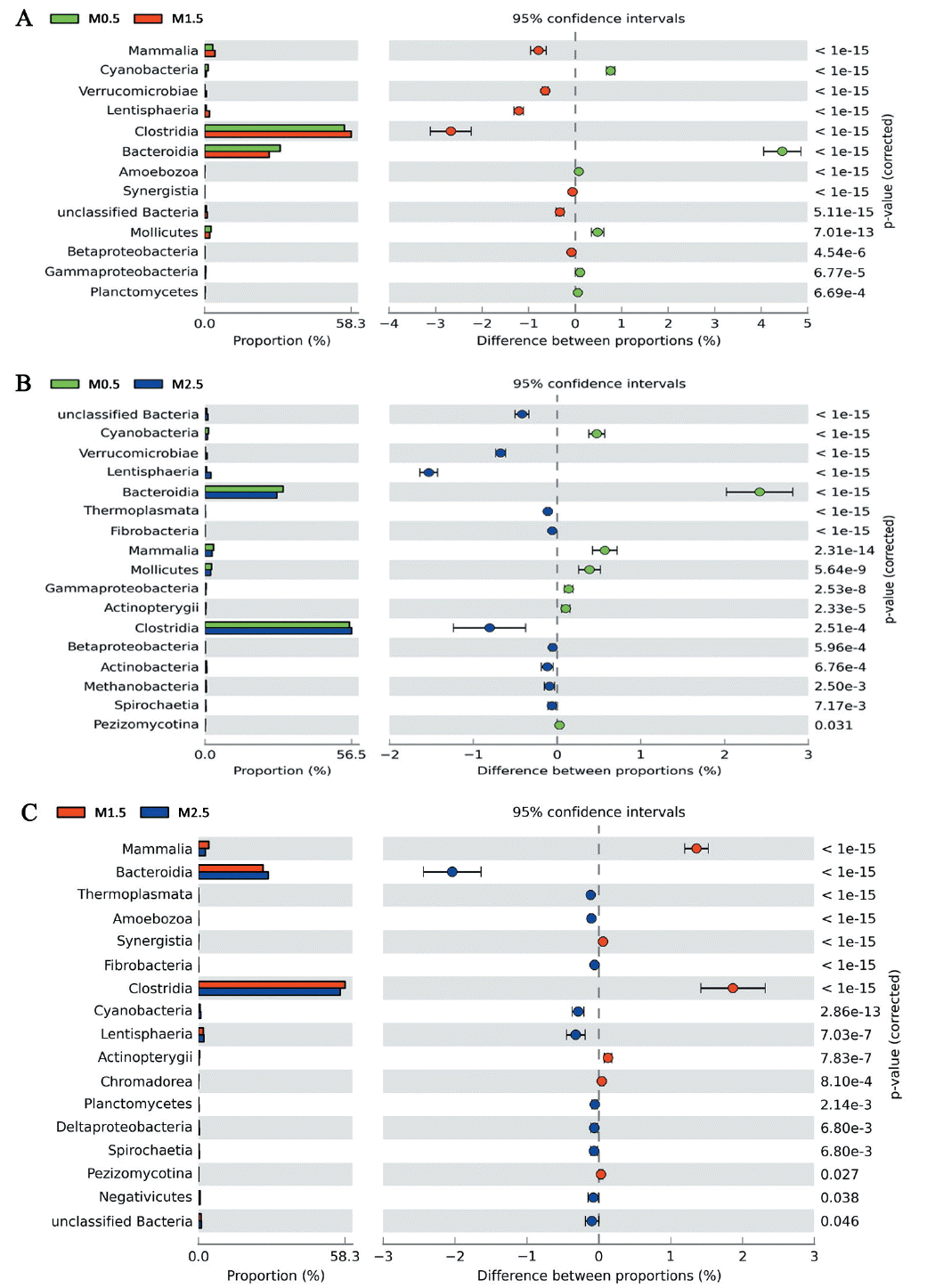
Deep metagenomic sequencing provides the opportunity to explore the existence of a common set of microbial species (common core) in the cohort. For this purpose, we aligned the Illumina GA reads of each yak gut microbial sample onto SILVA rRNA database, using a 1e-10 e-value, ≥60% identity threshold and a minimum of 50 bp alignment length, and determined the proportion of the genomes covered by the reads that aligned onto only a single position in the set. Phylogenetic classifications of the rRNA gene sequences are shown in Figure 2. As expected [5,19], more than 85% of rRNA gene sequences belong to the phylum Firmicutes and Bacteroidetes, including the most abundant gut species, such as the family Ruminococcaceae (46.5%), Rikenellaceae (11.3%), Lachnospiraceae (10.0%), and Bacteroidaceae (6.3%). Significant differences of gut microbial composition between pairwise comparison in yaks of 0.5, 1.5, and 2.5 years old by Fisher’s exact test are shown in Figure 3. The proportion changes of the class Clostridia, Bacteroidia, and Lentisphaeria (>1%) were more significant than that of others (<0.5%) in all age groups. Class Clostridia proportion increased first, and then decreased, with the increase of yaks’ age. Conversely, Class Bacteroidia proportion decreased in the young animals, and then increased with age. Class Lentisphaeria increased linearly with the increase of yaks’ age. These results are consistent with proportional change trend of the corresponding phylum Firmicutes, Bacteroidetes, and Lentisphaerae (Figure 2B). Other phyla, like Tenericutes and Cyanobacteria, decreased in the older animals (1.5 and 2.5 years old), while Proteobacteria had almost no changes during yaks’ growth. Compared with the young animals (0.5 years old), there were smaller proportional changes (<0.5%) of non-Clostridia, Bacteroidia, and Lentisphaeria animals from 1.5 years to 2.5 years (Figure 3).
Comparative functional analysis of the gut microbiome from yaks
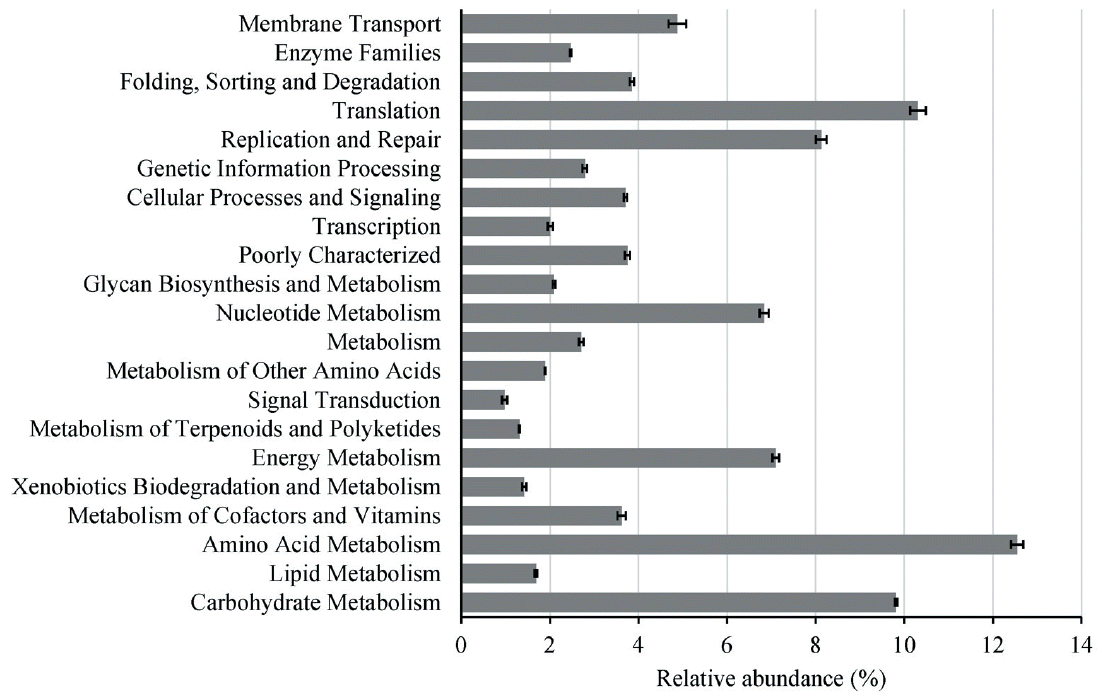
To identify the functions encoded by the gut genome in the yaks, we annotated the metagenomic sequences according to the KEGG using a 1e-10 e-value, ≥60% identity threshold and a minimum of 50 bp alignment length, and finally we identified a total of 6754 KEGG orthologous groups (KOs). Classifications of the KOs assigning to functional categories are shown in Figure 4, suggesting that the gut microbiome of yaks has enriched activity for metabolism of carbohydrates (9.8%), proteins (12.3%), amino acids (14.4%), nucleotides (6.8%), glycans (2.1%), lipids (1.7%), xenobiotics (1.4%), cofactors, and vitamins (3.6%). Moreover, pairwise comparative significant difference of gut microbial KEGG functional analysis of our data from the yaks of 0.5, 1.5, and 2.5 years old by Fisher’s exact test (Figure 5) demonstrated the most prominent differences involved diet-related processes such as energy metabolism, carbohydrate metabolism, amino acid metabolism, and metabolism of cofactors and vitamins, as well as xenobiotic-associated functions such as membrane transport and xenobiotic biodegradation and metabolism. Compared with the young animals (0.5 years old), there were smaller proportion changes (<0.5%) of these diet-related metabolism processes between 1.5- and 2.5-year groups (Figure 5).
Functional representation of gut microbiota in yaks
According to functional categories of the KOs identified, we found two types of functions among the range clusters: one is required in all bacteria (housekeeping) and another potentially specific for gut bacteria. Among the former category, most were related to the functions involved in the main metabolic pathways, e.g., central carbon metabolism, amino acid synthesis, as well as important protein complexes like RNA and DNA polymerase, ATP synthase, and general secretory apparatus. This is similar to the previous study in human gut microbiota [9].
Using gene functional classification in a metagenomic analysis of gut contents samples from yaks, we detected sequences homologous to genes coding 1,4-beta-cellobiosidase (EC 3.2.1.91) (46 genes), β-glucosidase (EC 3.2.1.21) (45,090 genes), 1,4-β-xylosidase (EC 3.2.1.37) (608 genes), and endo-1,4-β-xylanase (EC 3.2.1.8) (6,656 genes), which participate in the digestion of cellulose and hemicellulose, indicating that quite a few gut microbes, including the genus Clostridium, Lachnospira, and Ruminococcus, play important role in digesting cellulose in yaks. Furthermore, a variety of striking metabolic pathways seem crucial for the hosts, not unexpectedly, to biosynthesize essential amino acids by using the host diet and/or intestinal lining. Amino acids metabolic pathways were disclosed to be dominant in all yak gut samples by gene functional classification (Figure 4), which increased linearly with yaks’ age (Figure 5). Examples include amino acids metabolic pathways in the yaks’ gut microbiota (Table 1) for the synthesis of proteins in the hosts, such as sequences homologous to genes coding aminotransferase, synthase, kinase, ammonia-lyase, dehydrogenase, decarboxylase etc. that were related to the metabolism of aspartate (average 66,030 genes), glutamate (average 59,015 genes), serine (average 38,654 genes), glycine (average 28,529 genes), glutamine (average 25,206 genes) and so on, which surely make contribution to the normal growth of yaks.
DISCUSSION
Our current knowledge regarding the gut bacteria in yaks is very limited, contrasted that about the bacterial ecology and diversity in bovine rumen content and human gut. Although the bovine fecal microbiota has been well characterized by using culture-dependent methods, which are necessarily limited to characterize growing bacteria, while culture-independent methods can reveal community members recalcitrant to culture [20]. The PCR-DGGE approach had been previously used in studies of environmental ecology and microbial systems during food fermentation, as well as the studies of the populations present in the rumen or gastrointestinal contents [8]. However, because of the necessary steps of PCR amplification, the information regarding microbial community structure is limited. While metagenomic approaches based on the random sequencing of environmental DNA can provide a wealth of information (free of PCR bias) about gene content, metabolic potential, and the function of microbial communities [2,21]. In this study, we used PCR-DGGE and a connective way of metagenomic sequencing to study the structural and functional changes of gut microbial community during yaks’ growth to further understand probable benefits of gut microbiota on nutrient metabolism in hosts.
Recent research on gastrointestinal bacterial populations and host metabolic syndrome have sparked a renewed interest in the gut microbiome [22,23]. In mammals, dominant phyla were Firmicutes, Bacteroidetes, followed by Fusobacteria, Proteobacteria, Actinobacteria, but the proportion of each phylum fluctuated and was affected by multiple factors such as animal species [24]. It has been indicated that the microbial population of lower intestinal bacteria of cattle were dominated by strict anaerobes such as Bacteroides spp., Clostridium spp., and Bifidobacterium spp. [25]. These results supports findings from the current study where the predominant classes found in yaks were Clostridia, Bacteroidia, and Lentisphaeria, and the most abundant gut species were Clostridium, Bacteroides, Peptostreptococcus, and Ruminococcus in genus level. Clostridium spp. is a broad genus which is ubiquitous in the gastrointestinal tract, and Clostridia can influence the host both positively and negatively. Some Clostridium spp. may be beneficial to improve the digesting process of complex organic matter like cellulose and even act as beneficial probiotics [26]. Previous study on the giant panda’s gut microbiome reported that half of the predicted genes coding for cellulose and hemicellulose-digesting enzymes were found to be with Clostridium spp., like Clostridium butyricum [6]. Many Firmicutes spp. with demonstrated cellulose and hemicellulose degradation enzymes have been found in the intestinal tract, including members of the Lachnospiraceae and Clostridiaceae families, with a few from Eubacteriaceae and Bacillaceae families [27]. An additional niche that some Firmicutes, including Clostridium, Enterococcus, and Staphylococcus species, may fill in these enriched communities is metabolism of peptides and amino acids [28]. Bacteroides are well-known intestinal bacteria that can be both beneficial and harmful [29], and they are also noted to participate in natural genetic transfer of antimicrobial resistance genes [25]. Members of the Bacteroidaceae family have been demonstrated to be able to utilize a wide range of carbohydrates, including those that could form plant cell wall (cellulose, xylan, pectins, and β-glucans and galactans), host mucopolysaccharides and glycoproteins [27]. Thus, dominant microbes found in yaks’ gut, like the phylum Firmicutes, Bacteroidetes, and Lentisphaerae, the class Clostridia, Bacteroidia, and Lentisphaeria, as well as the genus Clostridium, Bacteroides, Peptostreptococcus, and Ruminococcus may be involved in the nutrient metabolism of the host, especially in the metabolism of carbohydrates, peptides and amino acids. Absolutely, this can be confirmed by the large number of genes discovered in yaks’ gut microbiota, which code enzymes related to the digestion of cellulose and hemicellulose and metabolism of amino acids. Dynamic changes of these gut microbiota may affect the nutrient absorption and utilization to yaks.
The gut microbiota can be impacted by numerous host factors, such as diet, age, antibiotic consumption, and the general host health. Moreover, these bacteria can be influenced by environmental factors, such as the geographical location, season, and feeding regimen [30]. Results of Ley et al [31] indicated that both host diet and phylogeny influence bacterial diversity, which increased from carnivory to omnivory and then to herbivory. However, the dynamic changes of gut microbiota of yak during growth were not well known. In this study, we first observed that gut microbial composition changed during yaks’ development by analysis of the DGGE fingerprint. Then, metagenomic results further to demonstrated that, the class Clostridia, Bacteroidia, and Lentisphaeria, as well as the phylum Firmicutes, Bacteroidetes, Lentisphaerae, Tenericutes, and Cyanobacteria, varied more during yaks’ growth, especially in young animals (0.5 and 1.5 years old). These changes might be influenced by the stress of weaning and shift in food composition, besides the age factor, as 0.5 years old of yaks were still not weaned, while yaks above one year old were fed on grass. In the weaning transition, the diet of yaks was changed from highly-digestible milk to less-digestible grass, which resulted in significantly increased proportions of Clostridia and Lentisphaeria, and decreased proportions of Bacteroidia, in older animals (1.5 years old). Large number of genes discovered in the genus Clostridium, Lachnospira, and Ruminococcus, which code enzymes related to the digestion of cellulose and hemicellulose, confirms the dynamic changes of the gut microbiota in older yaks. In view of the increasing proportions of Clostridia and Lentisphaeria, as well as the corresponding phylum Firmicutes and Lentisphaerae in older yaks’ gut while maintaining the same variation of amino acids metabolic pathways, indicates that these gut microbiota may participate in the metabolism of amino acids, thus furthers affects protein utilization and anabolism in yaks.
Because of the semi-open farming system and the lack of sufficient feed stuff in rangelands in winter and spring, some standardized rangelands have begun to feed supplementary silage forage to yaks in order to reduce animals’ high mortality in winter and spring. Thus, based on our study the beneficial bacteria in Clostridia, Bacteroidia, and Lentisphaeria, besides other genus, including Clostridium spp., Lactobacillus spp., and Bifidobacterium spp., are recommended to be added into feedstuff in order to control putrefying bacteria and further to regulate gut microbiota in the gut of weaning yaks. This strategy would improve the anabolism in yak intestine thus to increase yaks’ productivity, and related research will be carried out in the next step.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







