Genome-association analysis of Korean Holstein milk traits using genomic estimated breeding value
Article information
Abstract
Objective
Holsteins are known as the world’s highest-milk producing dairy cattle. The purpose of this study was to identify genetic regions strongly associated with milk traits (milk production, fat, and protein) using Korean Holstein data.
Methods
This study was performed using single nucleotide polymorphism (SNP) chip data (Illumina BovineSNP50 Beadchip) of 911 Korean Holstein individuals. We inferred each genomic estimated breeding values based on best linear unbiased prediction (BLUP) and ridge regression using BLUPF90 and R. We then performed a genome-wide association study and identified genetic regions related to milk traits.
Results
We identified 9, 6, and 17 significant genetic regions related to milk production, fat and protein, respectively. These genes are newly reported in the genetic association with milk traits of Holstein.
Conclusion
This study complements a recent Holstein genome-wide association studies that identified other SNPs and genes as the most significant variants. These results will help to expand the knowledge of the polygenic nature of milk production in Holsteins.
INTRODUCTION
Holsteins are the world’s highest-milk producing dairy cattle. Approximately 2,000 years ago, the black Batavians and white Friesians cows were bred to produce better breed. These cattle have been continuously selected and genetically evolved into the efficient, high producing black-and-white dairy cattle, which we know as Holstein-Friesian. For the last several decades, intensive application of traditional animal breeding technologies has significantly improved milk performance throughout the world.
Technology of molecular biology has opened up the possibility of identifying genome regions and variants underlying complex traits such as milk production, fat and protein. Unlike the traditional animal breeding programs which rely on phenotype and pedigree information, genetic evaluated information provide a great potential to enhance selection accuracies and expedite the genetic improvement of animal productivity. Since the seminal work on quantitative trait locus (QTL) mapping by Georges et al [1], a large number of articles including detection of QTLs for milk production traits have been published. So far, approximate 1,345 QTLs for milk production traits had been reported via genome scans based on marker-QTL linkage analyses. The limitations of QTL mapping using linkage analysis and/or linkage disequilibrium (LD) based on the panels of low to moderate density markers have been well documented [2].
The advent of genome-wide panels including hundreds of thousands of single nucleotide polymorphisms (SNPs) has resulted in the development of commercial SNP chips and rapid, large-scale genotyping of common SNPs across large populations. These SNPs have been widely used for the detection and localization of QTL for complex traits in many species [3], and have proved powerful and useful in identification of casual mutations associated with economically important traits in livestock [4,5] as well as human diseases. At the same time, genome-wide association studies (GWAS) based on high throughput SNP genotyping technologies open a broad avenue for exploring genes associated with milk production traits in dairy cattle [6]. Most recently, along with maturing of genome sequencing and high throughput SNP genotyping technologies, GWAS is becoming practical for exploring genes associated with complex traits. Like this, GWAS has been widely accepted as a primary approach for gene finding, and it achieved huge success in identifying genes conferring modest disease risks in human.
Several studies focusing on identifying genes for milk production traits had been performed. Associations between milk traits and polymorphisms in candidate genes have produced a long list of potential markers with significant effects reported in regional Holstein cattle population [7]. Generally, most of the economic traits in dairy cattle are controlled by polymorphisms, genes of small or large effects. To find genetic variant related to milk production traits beyond previous studies, we performed GWAS with genomic estimated breeding values (gEBVs) using 1,941 Korean Holsteins data. Estimated breeding value (EBV) was used as the phenotype as it only considers the genetic component of phenotypic variance. We then used p-value integration method to detect significant genetic regions with reduction of false positive error. Using this approach, we identified 9, 6, and 17 significant genetic regions associated with milk production, fat and protein, respectively and most of these genetic regions were not reported, previously. The identified genetic regions and their genes could be considered as a preliminary foundation for further studies in Holstein milk production traits. Furthermore, the identified genetic regions may be used as potential candidate markers for selection in Korean dairy cattle breeding programs and provide unprecedented insight into the structure of Holstein cattle populations.
MATERIALS AND METHODS
Animals and data
We used pedigree data containing 1,941 individuals (from present to 3 generations ago) in Korean Holstein population to infer EBV. There were milk traits record data (milk production, milk fat, milk protein) of 1,169 individuals of total 1,941. We took 911 individuals samples to perform SNP chip analysis and 488 individuals of 911 were overlapped with individuals of pedigree data. DNA was extracted from nasal discharge samples or semen of 911 Holstein individuals. DNA was quantified and genotyped using the Illumina BovineSNP50 BeadChip containing 54,609 SNPs. Features of the Illumina BovineSNP50 BeadChip have been detailed previously. All samples were genotyped using BEADSTUDIO (Illumina lnc, San Diego, CA, USA).
Genotype quality control and imputation
The chip includes 54,609 SNPs that are distributed on the 29 bovine autosomes, X and Y chromosomes with an average density of one SNP per approximately 49 kb) from the cow genome, UMD 3.1. We used three criteria to perform quality control to reduce false positive results. So, we excluded SNPs with Hardy–Weinberg equilibrium test p-value of <0.001, a missing rate of >0.05 and minor allele frequency of <0.01. Additionally, because we used cows and bulls in association analysis, SNPs on the X and Y chromosome were also excluded retaining finally 41,099 autosomal SNPs. The remaining 41,099 SNPs were distributed evenly on the autosomes (Supplementary Figure 1). These quality control processes were performed using the software PLINK [8]. The 41,099 autosomal SNP data of Holsteins after quality control was imputed without panels using BEAGLE [9].
Inferring estimated breeding value of milk traits
We inferred EBVs of parity 1 records of three milk traits (milk production, milk fat, milk protein), respectively. To improve the accuracy of the EBV, we consolidated the number of environmental factors by reducing the factors deemed unnecessary. Considering size of pedigree data and phenotype records data, we used season and year in inferring EBVs. A single-trait animal model was used to estimate the genetic parameters as EBVs. The animal model used in this study was as followed:
Where, y (n×1) was the vector of each milk traits, X (n×p) was the matrix of fixed effects (season and year in this study), Z (n×n) was the matrix of random effects (relationship matrix in this study), b and a were coefficients vector for X and Z, respectively and e (n×1) is the vector of residual error (meaning inexplicable factors). EBV is the coefficients vector of Z matrix. All parameters were estimated using the BLUPF90 program. We used parity 1 records of 1,941 individual in Korean Holstein population in this process and inferred EBVs of 1,941 individuals per each milk traits between 1990 and 2014. The fixed effects in this analysis were year and season (12 to 2: winter, 3 to 5: spring, 6 to 8: summer, 9 to 11: fall).
After inferring EBVs, we inferred gEBVs of 911 individuals which used to perform SNP analysis (milk production, milk fat and milk protein respectively). First, we estimated SNP effect of each trait using 488 individuals. The model of estimation SNP effect was as followed.
Where, y (n×1) was the vector of each milk traits EBVs, Z (n×p) was the matrix of SNP genotypes and e is the vector of the i.i.d. residual random error with e ~ N(0, Iσe2), where σe2 denotes a constant variance. a in this model is the coefficients vector for Z and marker effects of milk traits, simultaneously. We applied ridge regression to solve this model and we assigned ridge parameter (based on heritability of previous Korean Holstein population) to each model of milk traits (ridge parameter λ = σe2/σu2) [10].
In the ridge regression model, argmin (the argument of the minimum) meant that the set of points of the given argument for which the given function attained its minimum value in mathematics. And Z (n×p) is the matrix of SNP genotypes and a is the coefficient vector for Z. To check the accuracy of the solution of ridge regression, we used the 10-fold cross validation. Secondly, we estimated gEBV based on SNP effect, as follows:
All calculations in estimating gEBV were performed using R (“MASS” packages).
Genome-wide association analysis
We performed single association analysis using PLINK, as follows:
Where, y is a vector of each gEBVs of 911 genotyped individuals, x is each SNP information and b is coefficient value for x vector. After SNP association test, we used genomic control p-value instead of normal p-value. And we assigned integrated p-value to non-overlapped regions containing 5 SNPs to identify a significant genetic region instead of SNP. We performed p-value integration using R (“MADAM” packages). Genome-wide significance was defined based on genomic control p-value integration of 5 SNPs and Bonferroni method to correct p-value thresholds of significance after p-value integration: significant association of 0.05 false positives was used as a genome-wide significance. An overview of the results of test using Manhattan plots was produced by R. Because the SNPs were mapped on the UMD3.1 assembly, we used UMD3.1 gene information in Ensemble Genome Browser to investigate function of significant genetic region. We searched Ensemble gene ID and gene symbol which was overlapped with each regions. And then we assigned gene information to each regions. In this way, we identified relationship between each regions and animal trait through cow data of Animal QTL database.
RESULTS
Phenotypes used in this study were three traits related to milk of 1,169 Korean Holstein individuals (milk production, fat and protein) of parity 1. Milk production records were in range 3,473 kg to 13,734 kg. Mean and standard deviation were 8,730.68 kg and 1,481.864 kg, respectively. Milk fat records were in range 135 kg to 532 kg. Mean and standard deviation were 329.62 kg and 59.95 kg. In case of milk protein, records were in range 113 kg to 428 kg. Mean and standard deviation were 275.42 kg and 45.38 kg, respectively. All traits followed approximately normal distribution and their distributions are shown in Supplementary Figure 2. And all pairwise correlation of three traits were higher than 0.69 (milk production-fat: 0.69, milk production-protein: 0.925, milk fat-protein: 0.726) and their plots are shown in Supplementary Figure 3.
After quality control and imputation of 911 Korean Holstein individuals, we estimated gEBVs by two-step method. In first step, we estimated EBVs of each parity 1 milk traits using a single-trait animal model. We considered two fixed effect (season and year) in estimation EBVs and their relationships between traits and fixed effect are shown in Supplementary Figure 4 (season) and Supplementary Figure 5 (year). Through these relationship figures, we could identify that effect of year was more than season. We show EBVs distribution of each milk trait in Supplementary Figure 6. After estimation EBVs of 1,169 individuals (containing 488 SNP genotyped samples), we estimated the 41,099 SNP effect of each milk traits using 438 (training set of 10 fold cross validation strategy) of 488 individuals through ridge regression. SNP effect of each milk trait followed normal distribution and is shown in Supplementary Figure 7. We could estimate gEBVs through combining SNP genotyped information and estimated SNP effect. To test gEBVs accuracy, we compared EBVs with gEBVs using 49 individuals (test set of 10 fold cross validation strategy). The correlation coefficient of EBVs and gEBVs of each milk traits (milk production, fat and protein) of 49 individuals were 0.58, 0.70, and 0.68, respectively. Their correlation plots are shown in Supplementary Figure 8. In this way, we could estimate gEBVs of each three milk traits of 911 genotyped individuals and their distribution is shown in Supplementary Figure 9.
We compared the genotypes of 911 individuals with EBV as a phenotype, respectively. After performing single association analysis to these comparison, we identified that p-values from this analysis were the results of overestimation through an inflation factor much more than 1. The inflation factor of milk production, fat and protein were 2.19, 2.29, and 2.35, respectively. This was much higher than 1 and meant that using these p-values to identify significant genetic variants was not appropriate. This phenomenon is common in animal GWAS, because domesticated animals as Holsteins contain a massively structured population from small number of bulls and high linkage disequilibrium. To reduce false positive errors, we applied two methods to detecting significant genetic variants. One was that we used genomic control p-values instead of normal p-values. These genomic control p-values of each milk trait were calculated in PLINK. We identified that there were no inflation in genomic control p-values of all milk traits through Quantile-Quantile plots (shown in Supplementary Figure 10). Additionally, the inflation factor of milk production, fat and protein were 1.003471, 1.013479, and 1.011284, respectively. But none of the 41,099 SNPs exceeded the threshold of Bonferroni multiple test based on genomic control p-values in milk association test (genomic p-value< 1.21E-06, equivalent to p-value = 0.05 after Bonferroni multiple correction). Milk fat and protein association test results were same to Milk production (Supplementary Figure 11). And then we integrated genomic control p-values of five SNPs into one p-value through Fisher’s Method for combining p-values [11] and assigned a p-value to each region. We identified significant genetic regions which exceeded the threshold of Bonferroni multiple test based on integrated p-values (integrated p-values< 6.09E-06, equivalent to p-value = 0.05 after Bonferroni multiple correction). The results of region estimation in this GWAS study after chromosome sorting are in the Manhattan plots in Figure 1.
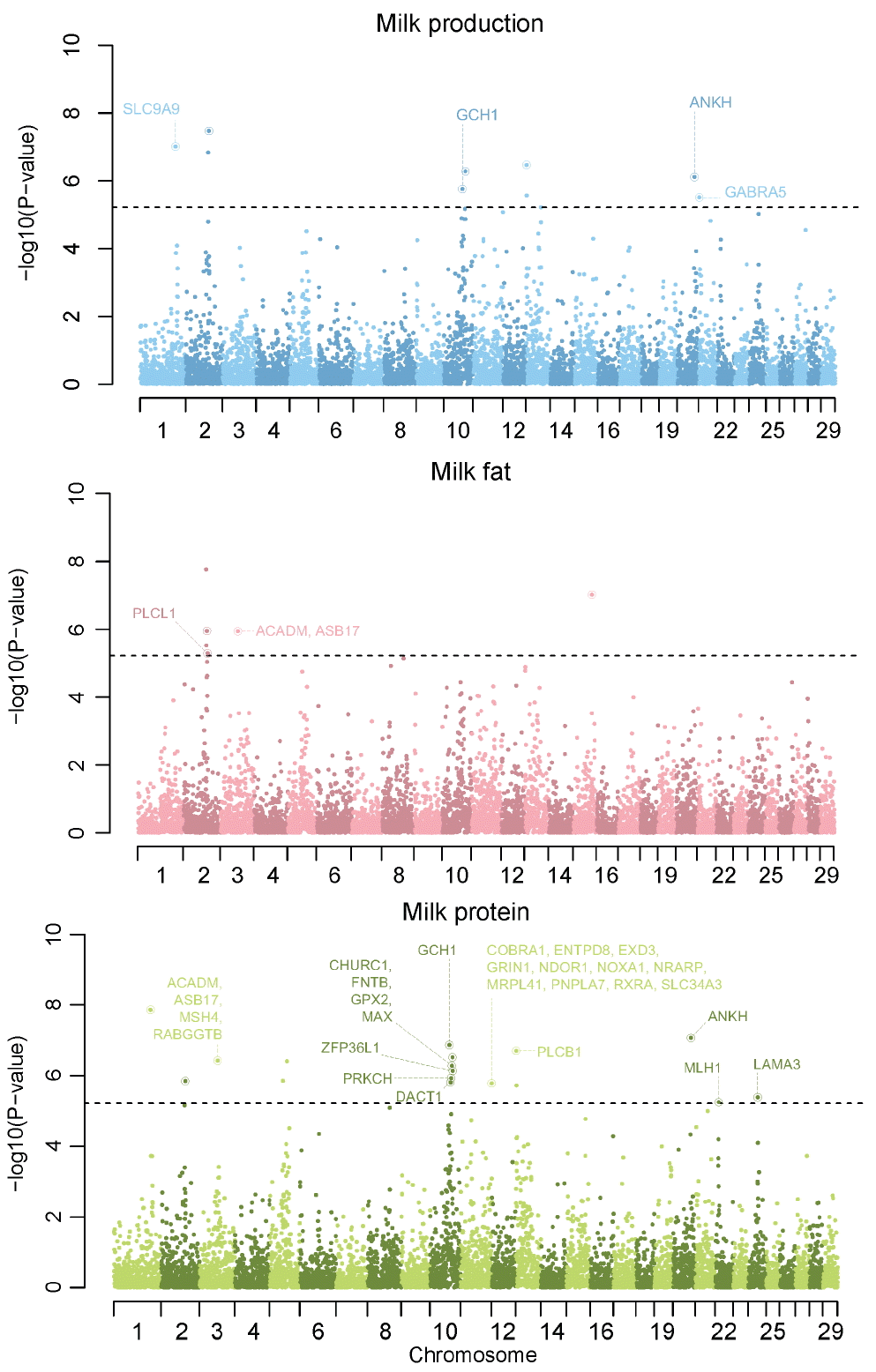
Manhattan plot of genome-wide association studies (GWAS) result of each milk traits after p-value integration. Each circle indicated each region containing 5 single nucleotide polymorphism (SNP) and circle with ring meant that region contained nearby gene. We provide the gene names which were significant in both GWAS and gene ontology analysis. a) milk production, b) milk fat, and c) milk protein. Grey dot line indicates the threshold of Bonferroni multiple test based on integrated p-values in each milk traits association test (genomic p-value<6.09E-06, equivalent to p-value = 0.05 after Bonferroni multiple correction).
In association test of milk production, nine regions (containing forty five SNPs) were significant and were distributed into six chromosomes (Table 1). Seven of nine regions had overlapped sixteen Ensemble genes ID and fourteen of sixteen Ensemble genes were related to the protein coding genes. All of the cow QTLs were related to nine significant regions in the association test of milk production. In association test of milk fat, six regions (containing thirty SNPs) were significant and were distributed into three chromosomes (Table 2). Four of six regions had overlapped thirteen Ensemble genes ID and twelve of thirteen Ensemble genes were related to 11 protein coding genes. Ten cow QTL were related to two of six significant regions in the association test of milk fat. Additionally, six of ten QTL were Holstein breed specific. In association test of milk protein, seventeen regions (containing eighty five SNPs) were significant and were distributed into ten chromosomes (Supplementary Table 1). Fifteen of seventeen regions had overlapped sixty two Ensemble genes ID and fifty seven of sixty two Ensemble genes were related to the protein coding genes. Twenty three cow QTLs were related to six of seventeen significant regions in the association test of milk fat. Additionally, seven of twenty three QTLs were Holstein breed specific. Diacylglycerol O-Acyltransferase 1 (DGAT1) was known as a major gene for milk traits in cow. DGAT1 is located in 1,795,351 bp to 1,804,562 bp of chromosome 14. Flanking markers of DGAT1 gene were ARS-BFGL-NGS-94706 (CHR14:1696470) and Hapmap52798-ss46526455 (CHR14: 1923292). Normal p-values of ARS-BFGL-NGS-94706 and Hapmap52798-ss46526455 were 0.04434 and 0.7297, respectively and did not passed criteria of Bonferroni multiple test. After p-value integration, the nearest genetic region of DGAT1 was located in 1,463,676 bp to 1,696,470 bp of chromosome 14. That region of which p-value was 0.136 and did not passed criteria of Bonferroni multiple test.

Significant genetic regions in GWAS results of milk production

Significant genetic regions in GWAS results of milk fat
After three association test, we compared each result with other results. The most interesting comparison result was that CHR2:80605588–81002535 was significant in all three association tests. In this region, there were 4 Ensemble genes and three were protein coding genes (TMEFF2, transmembrane protein with EGF-like and two follistatin-like domains 2; NABP1, nucleic acid binding protein 1; and SDPR, serum deprivation response). There were two significant regions in both milk production and milk fat. Seven regions were significant in both milk production and milk protein. And we found two significant overlapped regions in between milk fat and milk protein.
DISCUSSION
To identify genetic regions underlying milk traits of Korean Holstein, we performed GWAS with p-value integration in this study. These results were based on 911 genotyped Holsteins in Korea. Before the association study, we estimated 1,941 Korean Holsteins EBVs and we inferred 41,099 SNP effects of each milk traits. Using these SNP effects, we estimated gEBVs of 911 genotyped Holsteins. EBVs contains only the genetic effect of phenotype and we could predict genetic capacity of each individual based on the record of those individuals and their relatives. EBV was used to rank breeding stock for selection in animal breeding and we decided that EBV were appropriate dependent variables in this study. In gEBV estimation, we assumed that heritability of the three milk traits (milk production, milk fat, and milk proteins) were 0.23, 0.20, and 0.19, respectively. These heritabilities were reported in a previous study using Korean Holstein population [12].
We decided that gEBV was more proper than phenotype. The reason for this was bulls were more important than cows in animal breeding. But bulls do not have milk production trait data. However, if we used gEBVs as phenotype instead of traits data, we can performed GWAS using cows and bulls. Additionally, milk production traits are strongly affected by environmental factors (herd, season, and so forth). Also, sample size is very important factor in GWAS. If we used phenotypes in this study, our sample size was only 488 individuals. This sample size is insufficient, especially considering recent GWAS trends. If we used gEBV as phenotypes, we can use 911 individuals in this analysis and perform GWAS with alarger sample size. Before real animal capacity tests, gEBV was directly used as selection indicator. Superior dairy cattle were selected based on gEBV as result of genomic selection.
GWAS is a promising method to discover common genetic variants that could explain disease or interesting economic traits of animals and plants. But inflation always is a problem in domesticated animal GWAS. Lambda values of milk production, fat and protein were 2.190281, 2.299665, and 2.350455, which were much higher than expected. We guessed that a strong reason for this inflation was very large LD in the Korean Holstein population. Korean Holsteins have been under intense directional artificial selection to increase milk quantity and quality. This selection could reduce genetic diversity of Korean Holstein population and increase LD. A previous study reported a reduction genetic diversity of Korean Holstein population through the effective population size [13]. We thought that inflation of p-values in animal GWAS was a general phenomenon and applied appropriate methods to detect significant genetic variants. First, we used genomic control p-values which did not have an inflation problem. But we could not detect significant genetic variants after multiple test through Bonferroni correction (Supplementary Figure 11). The reason no SNP was significant in these association tests was because milk traits are complex phenotypes affected by several or many genetic factors instead of a few strong genetic factors. So we identified significant genetic regions associated with milk traits instead of SNPs. Also, we assumed that SNP was representative of a certain region and that a region test with the trait were repeated by SNPs in that region, because the Holstein LD block was very large. We defined that each region of the Holstein genome consisted of 5 SNPs and did not overlap. This meant that a continuous five SNPs on the physical map was one region in this study. We could define 8,209 regions on whole genome of Korean Holsteins and the mean and standard deviation of region size were 243,570.4 bp and 142,286.2 bp. We integrated 5 SNP p-values into 1 region p-values to assign significant level to each 8,209 regions. We thought that this approach could detect significant genetic regions and exclude false positive error. Using this approach, we identified several genetic regions and genes related to milk traits which have not been reported, previously.
CHR2:80605588–81002535 was significant in all three association tests and contained three protein coding genes (TMEFF2, NABP1, and SDPR). TMEFF2 encodes transmembrane protein with epidermal growth factor (EGF)-like and two follistatin-like domains 2. EGF was reported to affect various milk production traits [14]. NABP1 encodes Single-stranded DNA ssDNA-binding protein that is ubiquitous and essential for a variety of DNA metabolic processes, including replication, recombination, and detection and repair of damage. SDPR encodes a calcium-independent phospholipid-binding protein whose expression increases in serum-starved cells. Serum related to density of several substances in milk and these affected milk production. So, we guessed that these genes in CHR2:80605588–81002535 were strongly related to diverse mechanisms of milk production.
There were 14 significant protein coding genes in the milk production association test, and we performed gene ontology analysis using them. Four terms were significant and three of total four terms were related to ion transport (Figure 2). Solute carrier family 9, subfamily a member 9 (SLC9A9), ankylosis protein homolog (ANKH), and gamma-aminobutyric acid A receptor, alpha 5 (GABRA5) genes were in these ion transport terms. Kramer et al reported in a previous GWAS study that SLC9A9 was in a region with possible high influence on the observed milk production trait [15]. ANKH encodes a multi-pass transmembrane protein that controls pyrophosphate level and GABRA5 is one of GABA subunit which are ligand-gated chloride channels. Previous studies reported that ion balance was very important to Holstein lactation. For example, maintenance of calcium homeostasis is critical for many functions as hormone secretion and cation–anion difference affects health status and lactation performance [16]. Phospholipase C, Beta 1 (PLCB1) encoded by this gene plays an important role in the intracellular transduction of many extracellular signals. PLCB1 with GABRA5 were reported as significant mammary gland genes affected by level of nutrient intake in pre-weaned Holstein heifers [17].

Result of gene ontology analysis using gene sets of significant genetic regions as each genome-wide association studies (GWAS) result of milk production (skyblue), fat (pink), and protein (green).
There were 11 protein coding genes in milk fat association test, and we identified their biological meaning in Holsteins through gene ontology analysis. Two terms were significant and one of them was related to the lipid metabolic process (Figure 2). PLCL1 and ACADM (acyl-coenzyme A dehydrogenase, C-4 to C-12 straight chain) genes were in cluster of lipid metabolic processes. PLCL1 encodes a protein which is involved in an inositol phospholipid-based intracellular signaling cascade and a component in the phospho-dependent endocytosis process of GABA-A receptor. Also six QTLs (related to meat and carcass association: 3 QTLs, exterior association: 2 QTLs, milk association: 1 QTL) belonged to CHR2:86831095–87004473 region containing PLCL1 genes and three QTLs were Holstein specific [18]. Especially, a specific trait of milk association QTL (cattle QTL ID: 25003) was milk fat percentage and Holstein specific. ACADM is associated with lipid metabolism in fat depot and is the most important enzyme in the ACAD family [19]. Inside the mammary epithelial cell, the triglycerides synthesized at the outer surface of the smooth endoplasmic reticulum start coalescing and forming micro lipid droplets. Schlegel et al reported the relative mRNA abundances of ACADM genes involved in fatty acid oxidation in the liver of dairy cows in the transition period and at different stages of lactation [20]. Ran Zhang reported that low density lipoprotein receptor class A domain containing 3 (LDLRAD3) plays a central role in mammalian cholesterol metabolism through Next-Generation Sequencing in Transgenic Cattle [21]. Also four QTLs (related to reproduction: 3 QTLs, production: 1 QTL) belonged to CHR15:67269656–67429625 region containing LDLRAD3 genes and three QTLs related to reproduction were Holstein specific [22]. Additionally, specific traits of three reproduction QTLs (cattle QTL ID: 15,193, 15,194, and 15,192) were calving ease (direct), calving index and calving ease (maternal), respectively.
There were 54 significant protein coding genes in the milk protein association test, and we identified twenty gene ontology terms to detect biological meaning of 54 genes related to milk protein (Figure 2). We clustered twenty gene ontology terms into 7 main terms using hierarchical clustering method (Figure 3). Cluster 4 in Figure 3 had the most number of genes related to milk protein (21 genes) and biological meaning of this cluster was mRNA metabolic process. The need of energy and protein during lactation increases dramatically. In dairy cows there is more than a 5-fold increase in energy and protein requirements from late gestation to lactation [23]. Another study using more precise measurements of daily tissue protein synthesis reported that there is a 4-fold increase in mRNA translation in lactating compared to non-lactating mammary tissue in the cow [24]. Because the efficiency to transform dietary nitrogen into milk proteins is low (25% to 30%), protein synthesis is a highly active and energetically costly process, with only a minor part of the synthetic machinery apparently being used for production of milk proteins. Also previous study reported the abundance of the milk proteins (with the exception of albumin, as discussed below) is highly-dependent on the transcription level [25]. Cluster 5 in Figure 3 had 15 genes related to milk protein and biological meaning in this clustering was multicellular organism process. Multicellular organisms are composed of many specialized cells which differ in structure and function. So we guessed that these 15 genes have a special relationship with the milk protein ingredients or mechanisms. Interestingly, Cluster 3 in Figure 3 had 4 genes and their biological meaning was programmed cell death. Programmed cell death was not directly associated with milk protein. But programmed cell death has substantial meaning in Holstein mammary biological system. The regulation of cell death initiation coupled to the removal of cell corpses is an integral part of the mammary gland life cycle [26]. During pregnancy, epithelial cells of the mammary gland expand to form branched and lobuloalveolar structures to allow milk production after birth of the offspring. Then on weaning of the progeny, the mammary gland undergoes an important remodeling step, termed involution, during which the unessential mammary epithelial cells die and are largely removed [27]. Additionally, Baik reported that protein kinase C eta (PRKCH) were differential expressed in mammary tissues of lactating dairy cows [28].
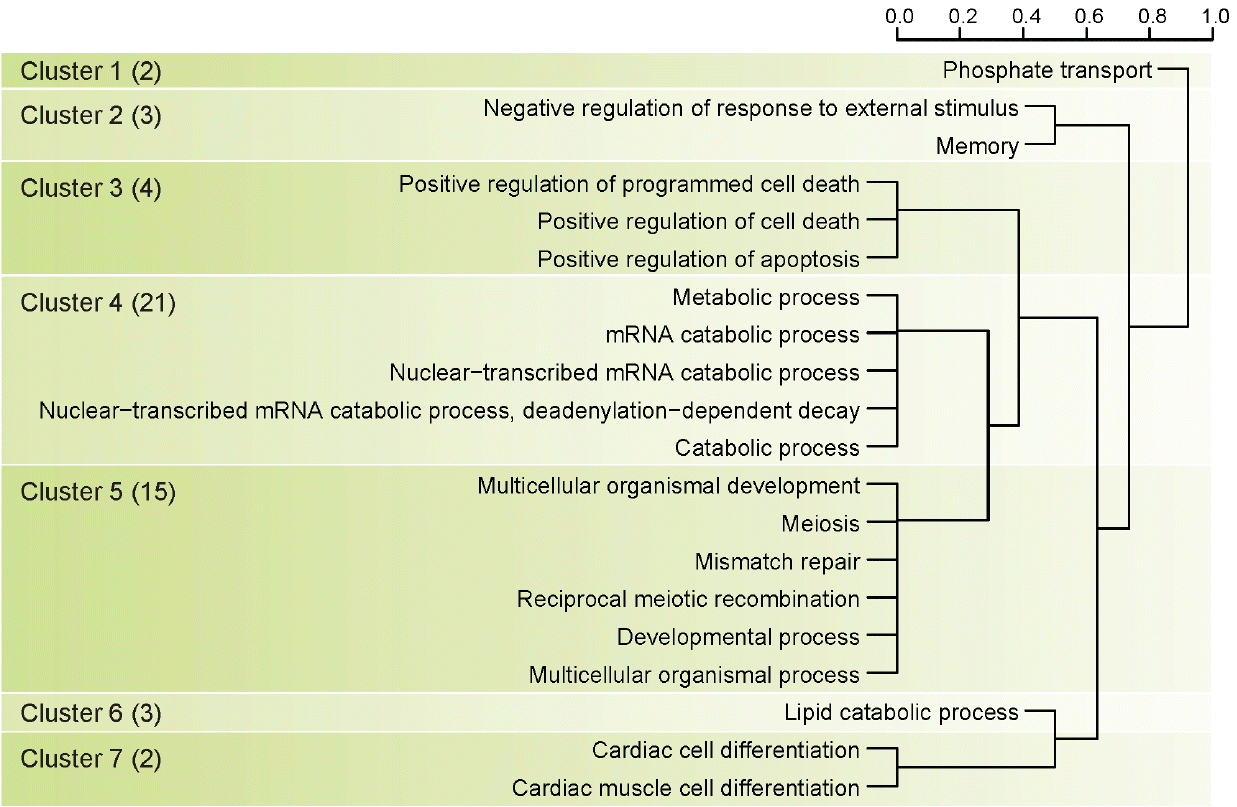
Clustering based on result of gene ontology analysis using gene set of significant genetic regions as genome-wide association studies (GWAS) result of milk protein. Number in parenthesis means number of genes in each cluster group.
Our results strongly support a major involvement of milk production in the genetic predisposition for increasing capacity of Holstein milk production and suggest several novel genes as genetic factors in milk production. Our results are not overlapped by other some previous GWAS of Holstein production traits. But several previous studies reported that some of our results were related to milk production traits of Holstein. Also, we identified that some of our results overlapped QTLs of cattle milk production. Although we will have to collect more samples and further research will be needed, we thought that our investigated genetic regions were biologically related to milk production traits.
CONCLUSION
These candidate regions and genes in our results may provide insight into the genetic makeup underlying milk production of Korean Holsteins.
Supplementary Information
ACKNOWLEDGMENTS
The work was supported by Project (PJ0092602015) of the National Livestock Research Institute of Rural Development Administration, Republic of Korea.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.