




INTRODUCTION
Necrotic enteritis (NE) is an economically important disease of the poultry industry worldwide, and is mediated by Clostridium perfringens strains that produce necrotic enteritis toxin B-like (NetB), a ╬▓-pore-forming toxin (Yan et al., 2013). NE was described in chicken for the first time by Parish in England in 1961 (Parish, 1961). The disease has been estimated to cost the world poultry industry roughly $2 billion annually (McReynolds et al., 2004). Over the past several years, several studies have profiled the gene expression of two genetically disparate inbred lines (resistant and susceptible line) with regard to their resistance or susceptibility against bacteria, protozoa and virus, including expression of Toll-like receptors (TLRs) and interleukin (IL) genes in response to Salmonella (Pan et al., 2011), beta-defensins in response to Eimeria maxima (Hong et al., 2012), and Th1 cytokines genes in response to Newcastle disease virus (Liu et al., 2012).
Recently, next-generation sequencing technology (RNA-Seq) has become available as a powerful tool to investigate transcriptional profiles for gene expression analysis of many organisms, such as Homo sapiens (Mortazavi et al., 2008), Chlamydomonas reinhardtii (Simon et al., 2013), Saccharomyces cerevisiae (Nagalakshmi et al., 2008), Fusarium graminearum (Zhao et al., 2013), Streptococcus mutans (Zeng et al., 2013), Catharanthus roseus (Van Moerkercke et al., 2013), mice (Cloonan et al., 2008), Arabidopsis (Lister et al., 2008) and Candida parapsilosis (Guida et al., 2011). RNA-Seq data has been shown to be efficiently used to improve gene model prediction, identify novel transcripts, and measure transcript expression in a single assay (Mortazavi et al., 2008; Trapnell et al., 2012). Furthermore, RNA-Seq technology is much more sensitive and efficient than the dedicated microarrays previously used to compare gene expression profiles (Mortazavi et al., 2008). RNA-Seq data also has been successfully used to identify alternative splicing in the genes of different species (Huang et al., 2013).
In addition, the different expression of miRNA were also examined in spleen and intestine of two chicken lines, namely, line 6.3 and line 7.2 which have been used in our previous small RNA next generation sequencing (NGS) studies (Hong et al., 2014). Herein, we used an RNA-Seq approach to perform transcriptome analysis in relation to the immune response in NE-afflicted chicken, and identified statistically significant gene expression differences of individual genes or transcripts between the spleens of two genetically disparate chicken lines. RNA-seq was carried out herein to identify mRNAs which are differentially expressed in the spleens of the two chicken lines experimentally afflicted with NE using co-infection with E. maxima and C. perfringens, and to gain an understanding of how immune related genes are regulated during NE for disease control in chicken.
MATERIALS AND METHODS
Experimental animal and necrotic enteritis disease model
White Leghorn chickens were used for the experiments, including the MarekŌĆÖs disease (MD) resistant line 6.3, and the susceptible line 7.2. These chicken lines have been selected and maintained for decades after exposure to avian leukosis virus and MD virus; line 6.3 being resistant, and line 7.2 being sensitive to these pathogens at the Avian Disease and Oncology Laboratory (ADOL), of the Agriculture Research Service, United States Department of Agriculture (USDA, East Lansing, MI, USA). Twenty chickens per group were randomly selected and infected with E. maxima strain 41A (1.0├Ś104 oocysts/chicken) by oral gavage on day 14, followed by oral gavage with C. perfringens strain Del-1 (1.0├Ś109 colony forming units [CFU]/chicken) 4 days later, as previous reported (Jang et al., 2012). The protocol for the development of NE was approved by the Beltsville Area Institutional Animal Care and Use Committee, USDA.
Total RNA preparation and quality control
On day 20 post-hatch, spleens were collected from five chickens per group. The spleens were carefully homogenized with a mortar and pestle after freezing with liquid nitrogen, and the total RNA from both lines was isolated using TRizol reagent (Invitrogen, Carlsbad, CA, USA) as described (Hong et al., 2012). RNA concentrations were quantified using a NanoDrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) at the wavelength of 260/280 nm ratio between 1.7 and 2.0. Integrity of the total RNA samples was evaluated using an Agilent 2100 (Agilent Technologies Inc., Santa Clara, CA, USA) and Tecan F2000 (Tecan Group Ltd., M├żnnedorf, Switzerland), and only the samples with RNA integrity number above 7.0 which were of high-quality (28S/18S >1) were used for the following experiments.
RNA-seq library construction, RNA sequencing and data analysis
RNA-seq library construction and RNA high-throughput sequencing were performed by Theragen Bio Institute (Suwon, Korea) using an Illumina HiSeq 2000 high throughput sequencer, according to the manufacturerŌĆÖs specifications. The RNA-Seq data were analyzed according to the method described (Trapnell et al., 2012). Briefly, reads were mapped to the Gallus gallus reference genome (v.4.0) obtained from the University of California, Santa Cruz (UCSC) database (UCSC: http://genome.ucsc.edu/) using TopHat v.2.0.3 (http://tophat.cbcb.umd.edu/), and Bowtie v.0.12.8 (http://bowtie-bio.sourceforge.net/index.shtml) from Illumina iGenomes (http://support.illumina.com/). Transcript abundance and differential expression were calculated with the Cufflinks program v.2.0.1 (http://cufflinks.cbcb.umd.edu/) as described (Trapnell et al., 2012). Levels of gene expression were normalized using values of the fragments per kilobase of exon per million mapped reads (FPKM). The following equation was used: RPKM = 109(C)/(N├ŚL), where C is the uniquely mapped counts determined from the high quality category, L is the length of the cDNA for the longest splice variant for a particular gene model, and N is the total mappable reads, which was determined as the sum of the high quality reads and the highly repetitive reads. Log2-transformations of the normalization were performed as specified in the methods described (Mortazavi et al., 2008), and the p-values calculated using the right-tailed Fisher exact test were <0.01. Subsequently, differential expression pattern analysis of the known mRNA and predicted novel mRNA was performed using unannotated sRNAs. Functional annotation in the form of GO was subsequently extracted from the database using Blast2GO v2.7.1 (http://www.geneontology.org/). The ŌĆ£TreeMapŌĆØ view of differences in the expression of GO terms was generated using REVIGO software (http://revigo.irb.hr/), as described (Supek et al., 2011). Further, the genes that significantly differed from the corresponding library were searched against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database to determine the pathways using DAVID Bioinformatics Resources version 6.7, NIAID/NIH (http://david.abcc.ncifcrf.gov/tools.jsp) with p<0.01.
Hierarchical cluster analysis for mRNA
Hierarchical cluster analysis of expressed genes was performed using Cluster software version 4.49 (http://www.bram.org/serf/Clusters.php) and Java Treeview software (http://jtreeview.sourceforge.net/). Cluster map analysis of the splenic genes detected between the two chicken lines was carried out using Euclidean distance. The p-values were calculated using the right-tailed FisherŌĆÖs exact test at the significance level of p<0.01.
Quantitative real-time PCR
For analysis of mRNA gene expression, 5 ╬╝g of total RNA were treated with 1.0 unit of DNase I and reverse transcribed using the Maxima first strand cDNA synthesis kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturerŌĆÖs recommendations. After cDNA synthesis, gene expression profiles were detected and quantified using an equivalent amount of cDNA. Briefly, cDNA (100 ng) was added to a reaction mix including 10 ╬╝L of 2├ŚPower SYBR Green Master Mix (Roche, IN, USA), 0.5 ╬╝L of each primer, and RNase-free water to a total volume of 20 ╬╝L. Real-time PCR (RT-PCR) was performed on a LightCycler 96 system (Roche) following the standard cycling program. Each analysis was performed in triplicate. Standard curves were generated using log10 diluted cDNA from individual and pooled total RNA, as described by Hong et al. Data from quantitative real-time PCR (qRT-PCR) were normalized relative to the expression of GAPDH using the 2ŌłÆ╬ö╬öCt method, as previously described (Livak and Schmittgen, 2001). Oligonucleotide primers for analysis and the GAPDH control were designed based on sequences available in public databases using Lasergene software (DNASTAR Inc., Madison, WI, USA) as indicated in Table 1.
Statistical analysis
Statistical analyses were performed with IBM SPSS ver. 20 (IBM Co., Armonk, NY, USA). The data were expressed as the mean┬▒SD for each group (N = 5), and comparisons between groups were carried out using Student t-test. Statistical significance was defined as p<0.05 and p = 0.01.
RESULTS
RNA sequence alignment
A total of 42.7 and 42.6 million sequence reads for chicken lines 6.3 and 7.2 were obtained by RNA-seq as indicated in Table 2. Of the total reads, 79.15% and 78.50% were successfully mapped to the chicken genome in lines 6.3 and 7.2, respectively. In each group, 57.11% and 56.57% of the sequences were properly paired, respectively. The percentage of properly paired sequences was not higher because only the reads aligned entirely inside exonic regions were included, and 54.77% and 52.49% of the reads were mapped to intergenic/exon-intron regions in the spleens of lines 6.3 and 7.2, respectively. Overall, line 6.3 had a higher number of successfully mapped reads than line 7.2 for the spleen.
Based on the number of sequence reads, the expression levels of the genes in each sample can be estimated. These fragments or reads were used to measure the levels of gene expression, and to identify novel splice variants of genes. As a result, the mRNA of 29,997 genes was detected, which was composed of 14,479 (49%) known genes and 15,518 (51%) novel genes.
To evaluate the quality of the RNA-Seq data, several quality control analyses were performed. In this study, gene coverage was used for evaluation of RNA-seq. Gene coverage is defined as the percentage of a gene covered by reads, and is equal to the ratio of the base number in a gene covered by unique mapping reads to the total base numbers of that gene. As shown in Supplementary Figure S1, the distribution of distinct reads over different read abundance categories showed similar patterns in the RNA-Seq libraries of the two chicken lines. In both NE-afflicted chicken lines, the read coverage was more than 90% of the chicken genome for 67% of the read sequences in both NE-afflicted chicken lines and 70% of the chicken genome for 12% of the read sequences.
Gene ontology analysis
Gene ontology (GO) plays an important role in the annotation and categorization of sequences, including unidentified and unannotated sequences. GO enrichment analysis provides all GO terms enriched significantly in the differentially expressed genes (DEGs) compared to the genome background, and filters the DEGs which correspond to biological functions. This method maps all DEGs to GO terms in the database, calculating gene numbers for every term, then uses a hypergeometric test to find the GO terms which are significantly enriched in the DEGs compared to the genome background with p<0.01, fold change Ōēź2, and then the ŌĆ£TreeMapŌĆØ view can be used to visualize the differentially expressed GO terms using REVIGO software.
In chicken line 6.3, 88 GO molecular functions were revealed to be annotated for 313 genes, in which 244 of the genes were upregulated and 69 genes were downregulated. In addition, 135 GO biological processes had annotations for 249 genes, of which 153 were upregulated and 96 were downregulated. For 22 GO cellular components which contained 176 genes, 134 of the genes were upregulated while 42 were downregulated (Table 3 and Supplementary Table S1a). On the other hand, chicken line 7.2 had 54 GO molecular functions for which there were 108 genes annotated genes: 67 upregulated and 41 downregulated. Of 70 GO biological processes annotated with 127 genes, 68 genes were upregulated while 59 were downregulated. 10 GO cellular components containing 29 genes, of which 12 were upregulated and 7 were downregulated (Table 3 and Supplementary Table S1b).
When NE-afflicted MD-resistant chicken line 6.3 was compared to the MD-susceptible line 7.2, the following GO annotations were revealed: 76 GO molecular functions were annotated with 239 genes, of which 149 were upregulated and 90 were downregulated; 141 GO biological processes were annotated with 234 genes, of which 83 were upregulated and 151 were downregulated; and 34 GO cellular components were annotated with 221 genes, of which 134 were upregulated and 87 were downregulated (Table 3 and Supplementary Table S1c). Overall, the number of genes up and downregulated in the spleen of line 6.3 was higher than in line 7.2. The NE-afflicted line 6.3 contained more upregulated genes in the molecular function and cellular component categories than line 7.2, whereas the inverse was observed in biological processes, as summarized in Table 3.
The four major categories clustered in the biological process ontology of chicken line 6.3 were signal cellular histone metabolism, positive regulation of polarized epithelial cell differentiation, nitrogen compound transport and cellular response to cell-matrix adhesion. In the spleen of line 7.2, most of the DEG categories in the biological process were related with arginine metabolism and ion transport (Figure 1). However, two major clusters of the cellular components in chicken line 6.3 were focused intrinsic to the membrane and extracellular space, and the major components of four clusters fell into lipid particles, outer mucus layer, anchored to the membrane, and membrane in line 7.2 (Figure 1). On the other hand, molecular functions for metallopeptidase activity, transmembrane transport activity and calcium ion binding were also clustered in line 6.3, while four major clusters indicated lytic transglycosylase activity, bile acid activity, short chain carboxylesterase activity and phosphoendopyruvate carbonxykinase activity in line 7.2 (Figure 1).
Identification of differentially expressed genes in two chicken lines
Generally, the number of mRNA-seq reads generated from a transcript is directly proportional to that transcriptŌĆÖs relative abundance in the samples (Trapnell et al., 2012). In this study, DEGs of chicken lines 6.3 and 7.2 were compared with p<0.01 and fold change Ōēź2.
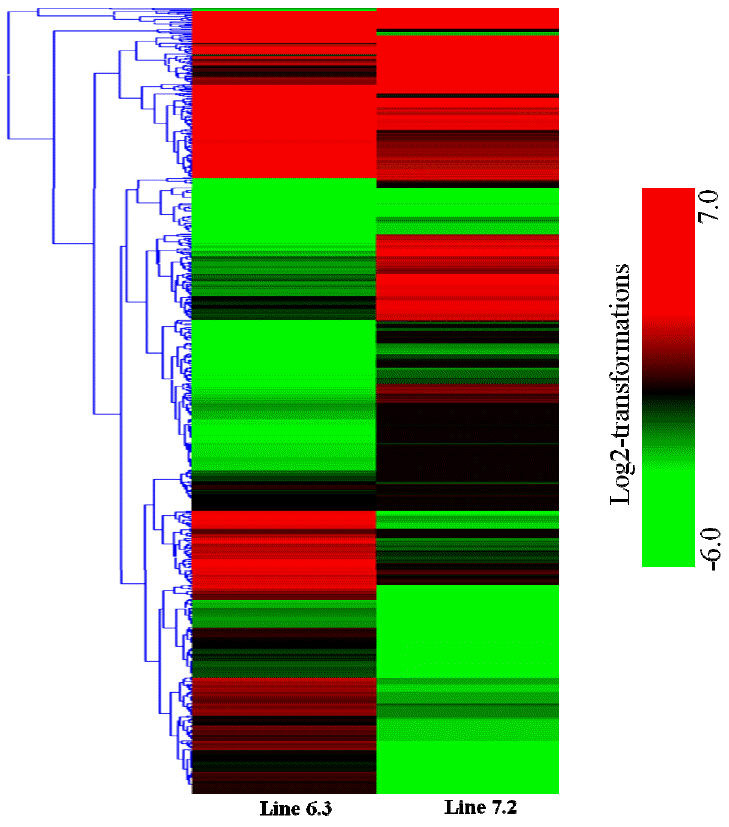
First, the gene expression profiles of the two NE-afflicted chicken lines were examined. The results indicated that 2,163 genes were significantly upregulated while 1,092 genes were downregulated in chicken line 6.3, with p<0.01 and fold change Ōēź2, whereas 1,388 genes were significantly upregulated and 1,080 genes were downregulated in line 7.2 (Table 2). In addition, comparison of the spleen expression files of the two NE-afflicted chickens revealed 2,234 genes to be significantly expressed between the two NE-afflicted chicken lines. Of the 2,234 genes, 1,239 were abundantly expressed in chicken line 6.3 compared to line 7.2, and 995 were highly expressed in line 7.2 with p<0.01 and fold change Ōēź2 (Table 2 and Figure 2).
To understand the functions of the DEGs, we mapped them using the Kyoto Encyclopedia of Genes and Genomes databases (KEGG) (http://www.genome.jp/kegg/) for signalling pathways analysis based on using DAVID Bioinformatics Resources version 6.7 with p<0.01. In KEGG pathway mapping, 1,311 genes could be assigned gene identification number with the National Center for Biotechnology Information (NCBI Gene ID). We identified 1,129 and 1,165 DEGs in chicken line 6.3 and line 7.2, respectively that could be mapped into various pathways at the KEGG (data not shown). The top 14 most differentially expressed signalling pathways from each comparison group are listed in Table 4. Specifically, the number of up and downregulated genes involved in mitogen activated protein kinase (MAPK) signalling pathway, endocytosis signalling pathway, cytokine-cytokine receptor interaction, JAK-STAT signalling pathway, Toll like receptor pathway, focal adhesion, erythroblastic leukemia viral oncogene homolog (ErbB) signalling pathway and transforming growth factor beta (TGF-╬▓) signalling pathway were significantly represented the largest group, which may have an important role in response of spleen in two chicken lines to the E. maxima/C. perfringens (EM/CP) infections.
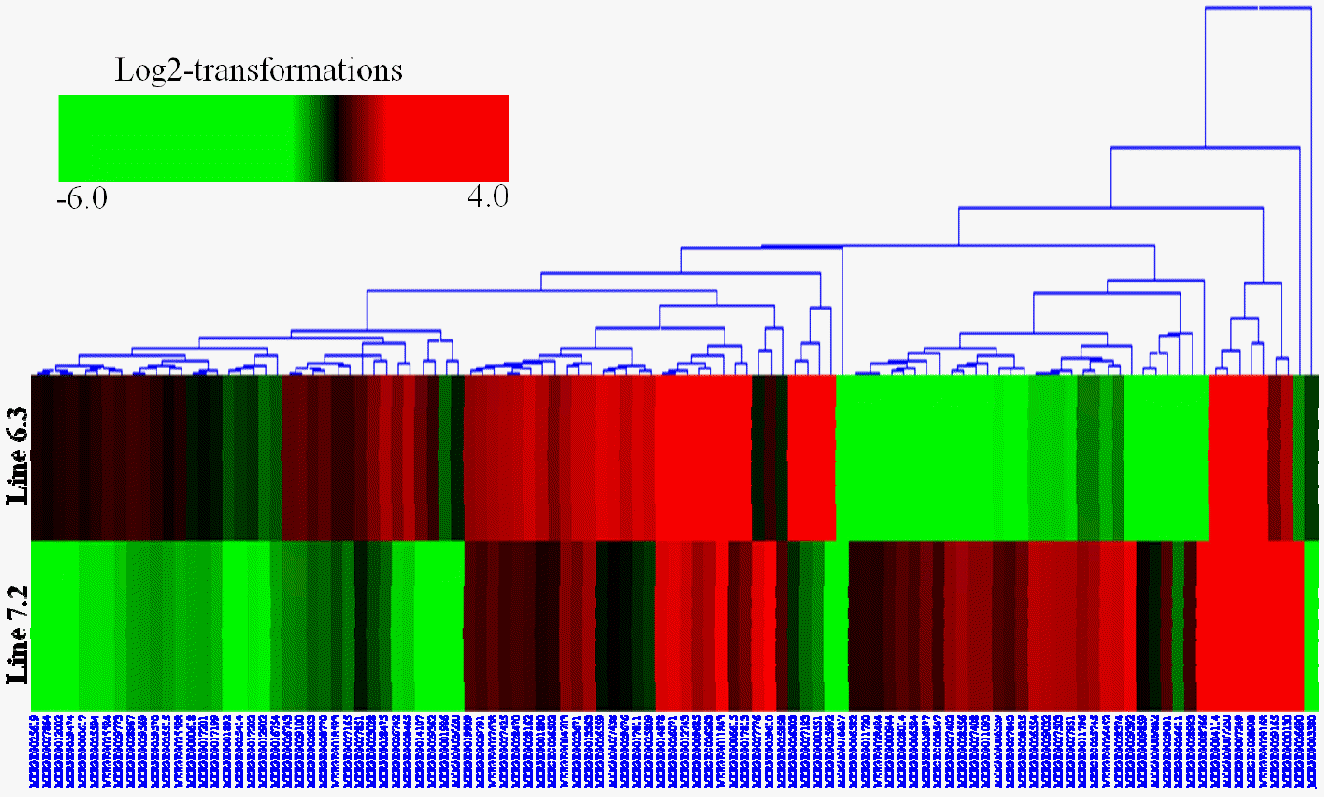
Hierarchical clustering was then performed for the analysis of 107 novel genes (Supplementary Table S2) found to be differentially expressed in the spleens between the two NE-afflicted chicken lines using Euclidean distance. In Figure 3, red indicates the upregulated and green the downregulated mRNAs between the two lines. Over half of the genes showed higher expression in resistant chicken line 6.3 than in susceptible line 7.2. Among the novel genes of interest, many were associated with response to stress, cellular, binding, membrane and adenosine triphosphate (ATP) activity. A number of the transcripts have specific involvement in the immune response.
Immune-related genes
Immune-related genes are biologically important for the host response to pathogens. Due to differences in the genetic background between chicken line 6.3 and line 7.2, it was expected that some immune related genes would be differentially expressed between the two lines of chicken. The data revealed a total of 139 immune related genes among the samples, with 130 genes in chicken line 6.3 and 117 genes in line 7.2 that were upregulated at p<0.01, and fold change Ōēź2. Comparison of chicken line 6.3 to line 7.2 indicated 12 immune related genes to be expressed at higher level in line 7.2 than line 6.3, whereas 18 immune related genes were more abundantly expressed in line 6.3 than line 7.2. Among the large number of DEGs, most of the up- and downregulated genes were related to protein transportation, modification and degradation in immunization. These genes are likely related to the degradation and processing of antigens for major histocompatibility complex (MHC) class I and II molecules. Most of the DEGs in the spleen were related to MHC-II antigen processing pathways, such as immunoglobulin, interferon, interleukin, heat shock protein 70 (Hsp70), and hemoglobulin (Supplementary Table S3). Moreover, we mapped all of DEGs immune-related genes in two chicken lines to the KEGG database for signalling pathways analysis and particularly, those involved in cell adhesion molecules pathway, Integrin signalling pathway and JAK/STAT signalling pathway.
Innate immune responses in necrotic enteritis-afflicted chicken lines
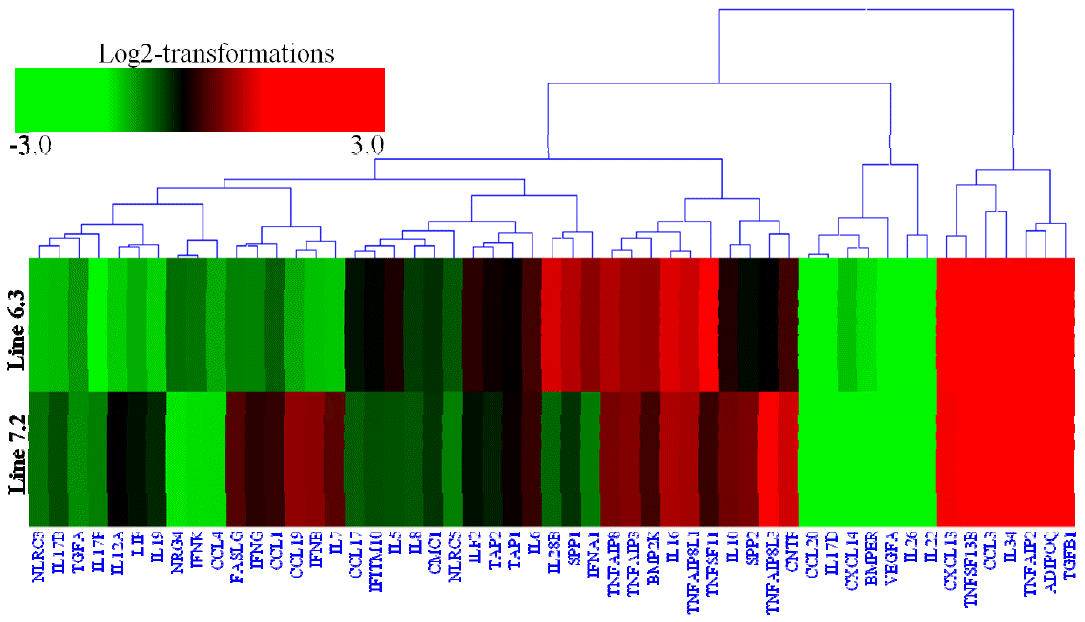
Transcription analysis of 53 cytokines and 97 cytokine receptors of the two NE-afflicted chicken lines are shown in Supplementary Table S4. The expression of 15 cytokines genes (ADIPOQ, BMP2K, CCL3, CXCL13, IL-16, IL-28B, IL-34, SPP1, TGFB1, TNFAIP2, TNFAIP3, TNFAIP8, TNFAIP8L1, TNFSF11, and TNFSF13B) was significantly upregulated by 2.1- to 9.25-fold (Supplementary Table S4, Figure 4), while 46 cytokine receptors (CSF3R, CXCR7, IL-17RA, IL-18R1, IL-1R1, IL-1R2, IL-20RA, IL-21R, IL-2RG, IL-4I1, IL-5RA, IL-7R, IL-9R, LILRB3, TGFBR1, TLR1, TLR15, TLR21, TLR5, TLR6, TLR7, TNFRSF13B, TNFRSF13C, TNFRSF1B, TRAT1, XCR1, etc.) were also significantly upregulated by 2.04- to 10.16-fold in chicken line 6.3 with p<0.01 and fold Ōēź2 (Supplementary Table S4, Figure 5). In contrast, 18 cytokines genes (BMPER, CCL19, CCL20, CCL4, CXCL14, IFNB, IL-12A, IL-17B, IL-17D, IL-17F, IL-19, IL-22, IL-26, IL-7, LIF, NLRC3, TGFA, and VEGFA) and 14 receptor genes (CXCR3, IFITM5, IL-13RA1, IL-17RC, IL-1RAPL2, IL-22RA1, IL-22RA2, ILDR1, LILRA5, LTBP2, PDGFD, PDGFRL, TNFRSF14, and TNFRSF6B) were markedly downregulated by 2.05- to 7.48-fold and 2.1- to 10.1-fold, respectively, in chicken line 6.3 with p<0.01 and fold Ōēź2 (Supplementary Table S4, Figures 4 and 5).
Similarly, the expression of 18 cytokine genes (IL-7, IL-10, TNFAIP8, SPP2, TNFAIP3, IFNB, CCL19, TNFAIP8L1, IL-16, CNTF, CXCL13, TNFAIP8L3, TNFSF13B, IL-34, TGFB1, TNFAIP2, CCL3, and ADIPOQ) and 39 cytokine receptor genes (CXCR7, CSF2RA, IL-1RL1, I-L1R2, IL-17RA, IFNAR1, IL-18RAP, IL-31RA, IL-9R, IL-18R1, IL-20RB, IL-21R, IL-2RG, IL-20RA, IL-4I1, LILRB1, TLR1, IL-17REL, LILRB5, TGFBR1, TLR6, TNFRSF13B, XCR1, TLR7, TNFRSF13C, TLR15 etc.) was also markedly upregulated by 2.03- to 8.41-fold and 2.08- to 9.12-fold, respectively, in chicken line 7.2 with p<0.01 and fold Ōēź2 (Supplementary Table S4, Figures 4 and 5). In addition, the expression of 7 cytokines genes (IL-22, VEGFA, IL26, CXCL14, BMPER, IL-17D, and CCL20) and 16 receptor genes (IL-22RA1, CXCR3, IL-17RC, IL-1RAPL2, IFITM5, NRG2, TNFRSF14, LEPREL2, IL-11RA, LTBP2, ILDR1, IL-17RD, MC1R, IL-22RA2, LEPREL1, and LILRA5) were significantly downregulated by 3.6- to 5.7-fold and 2.14- to 9.12-fold, respectively with p<0.01 and fold Ōēź2 (Figures 4 and 5).
When the cytokine genes were compared between the two lines, 6 genes (CXCL13, IL-28B, IFNA1, TNFSF11, SPP1, and TNFSF13B) showed higher expression in line 6.3, while the gene expression levels of 16 genes (IL-10, SPP2, CMC1, IL-19, LIF, TAP1, IFNG, IL-6, FASLG, IL-7, CCL19, TNFAIP8L3, IL-34, CCL3, IL-10, and SPP2) were higher in line 7.2 (Supplementary Table S4, Figure 4). Of the cytokine receptors, 20 showed significantly higher expression in line 6.3, while 8 were more abundantly expressed in line 7.2 with p<0.01 and fold Ōēź2 (Supplementary Table S4, Figure 5).
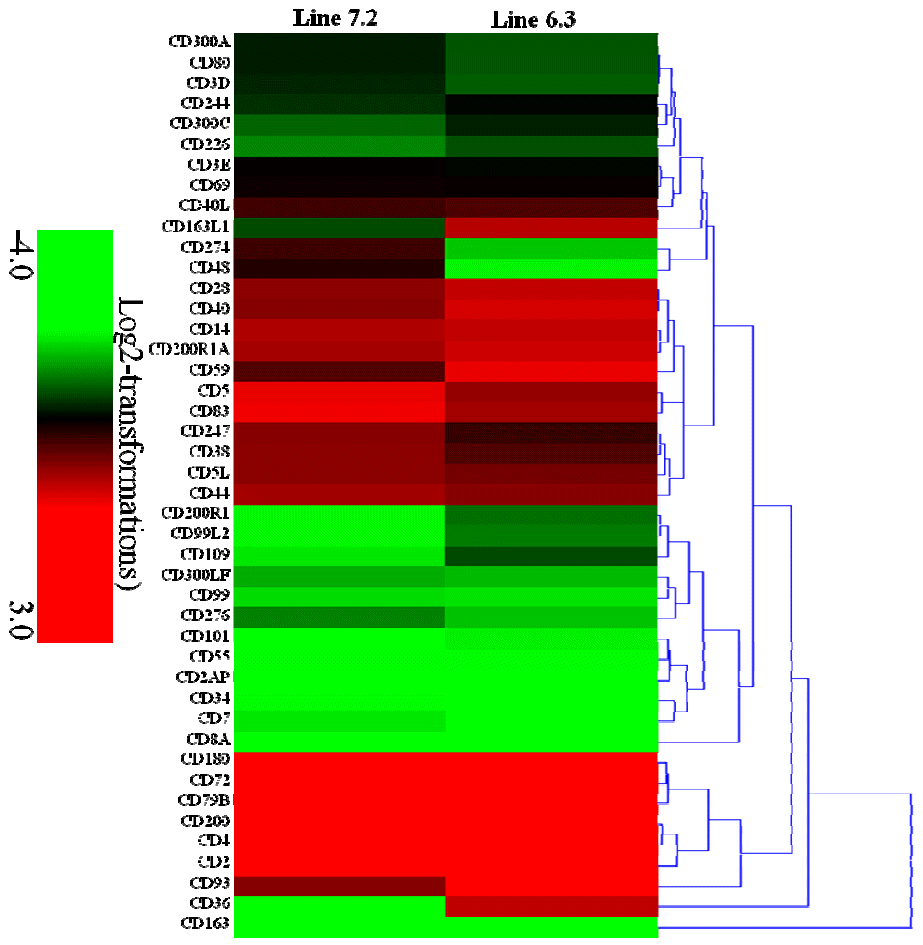
Transcription analysis of 44 CD genes was also carried out, indicating 24 CD genes (CD3E, CD244, CD69, CD247, CD40L, CD38, CD5L, CD44, CD5, CD83, CD163L1, CD36, CD14, CD28, CD200R1A, CD40, CD59, CD200, CD4, CD2, CD72, CD180, CD79B, and CD93) to be markedly increased by 2.1- to 8.4-fold, and 4 CD genes (CD163, CD8A, CD34, and CD7) to be significantly decreased by 2.1- to 8.2-fold in chicken line 6.3 with p<0.01 and fold Ōēź2. In contrast, 23 CD genes (CD3E, CD69, CD48, CD274, CD40L, CD59, CD40, CD93, CD247, CD28, CD5L, CD38, CD44, CD200R1A, CD14, CD5, CD83, CD2, CD200, CD4, CD79B, CD72, and CD180) were significantly increased by 2.1- to 8.7-fold while 2 CD genes (CD163 and CD36) were significantly decreased by 5.0- and 6.8-fold in chicken line, 7.2 with p<0.01 and fold Ōēź2 (Supplementary Table S5, Figure 6).
Discovery of novel genes in two chicken lines
For exploration of novel mRNA transcripts, the RNA-seq data were used to identify novel spliced transcripts consisting of novel splicing events of known annotated exons. Several programs are available for such analysis, including Qpalma, Splicemap, Mapsplice, TopHat, GSNAP and PALMapper, in which the TopHat algorithm pairs candidate exons and evaluates the alignment of reads to such candidates (Trapnell et al., 2009). In this study, mRNA for 29,997 genes was detected including 15,518 novel genes. Among the total novel genes, 3,700 and 3,407 were found to be upregulated in chicken lines 6.3 and 7.2, respectively with p<0.01 and fold Ōēź2. Moreover, comparison of the individual genes between the lines showed that the number of individual genes differentially expressed was generally equal in the NE-afflicted chicken line 6.3 (810 novel genes) and chicken line 7.2 (813 novel genes), with p<0.01 and fold Ōēź2. In addition, comparison of the expression of candidate novel genes between line 6.3 and line 7.2 revealed 1,503 and 1,047 novel genes to be significantly upregulated in chicken lines 6.3 and 7.2, respectively, with p<0.01 and fold Ōēź2 (data not shown).
Quantitative real-time PCR for DEG
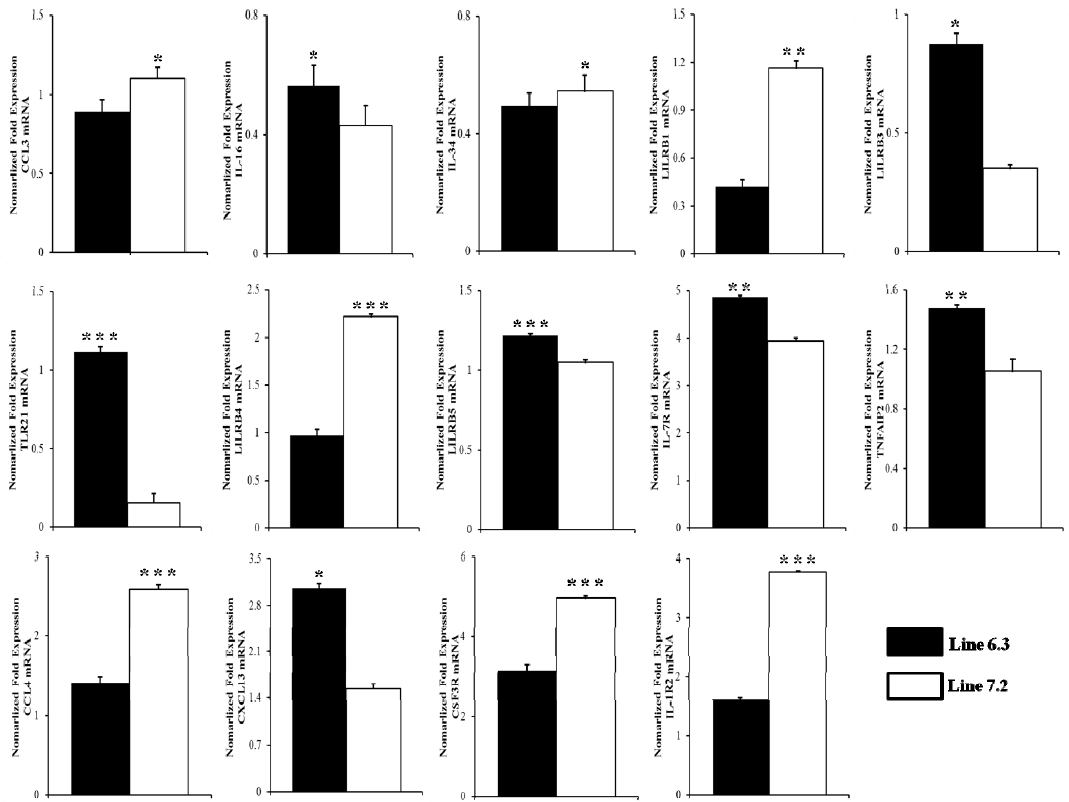
In order to validate the DEGs identified by RNA-Seq, 14 genes were selected for analysis via qRT-PCR. The genes were selected based on functional enrichment and pathway results from those with differing expression patterns. Primers were designed based on available sequences to amplify the specific altered genes. Primer sequences and the sizes of expected products are shown in Table 1. Fold changes from qRT-PCR were compared between the two chicken lines in Figure 7 and Supplementary Figure S2.
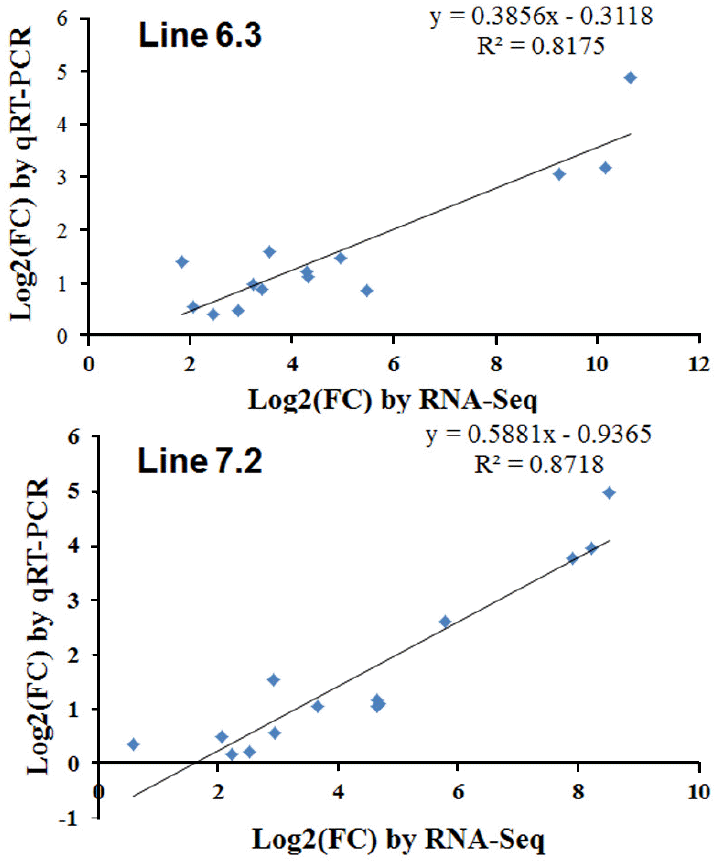
The qPCR results revealed consistent expression patterns with the differential expression observed by RNA-Seq analysis for all 14 genes. The expression trends were consistent for all transcripts in both analyses, with a correlation coefficient of R2 = 0.81 for chicken line 6.3 and R2 = 0.87 for chicken line 7.2 line 7.2 as shown in Figure 8. The expression of seven genes (IL-16, LILRB3, TLR21, LILRB5, IL-7R, TNFAIP2, and CXCL13) was significantly higher in chicken line 6.3 compared to line 7.2, while the other seven genes (CCL3, CCL4, CSF3R, IL-34, LILRB1, LILRB4, and IL-1R2) showed significantly higher expression in chicken line 7.2 than line 6.3 (Figures 7 and Supplementary Figure S2).
DISCUSSION
High throughput RNA-Sequencing technology is a recently developed approach to transcriptome profiling using deep-sequencing technologies and generates concomitant gene expression and polymorphism data over the whole transcriptome through a single experiment (Ozsolak and Milos, 2011). NE disease mainly targets in the intestine and several publications demonstrated that the level of mRNA expression were significantly regulated in intestine of two chicken lines coinfected with E. maxima and C. perfringenes, in which the expression of mRNA was highly increased in chicken line 6.3 than chicken line 7.2 (Kim et al., 2014) but the global mRNA expression in spleen of two chicken lines coinfected by E. maxima and C. perfringenes is not investigated yet. On the other hand, the spleen is involved in both the humoral and cellular immune responses through its role in the generation, maturation and storage of lymphocytes (Redmond et al., 2010). Gene expression study of the spleen of chicken is commonly used as an indicator of immune response (Redmond et al., 2010). In the present study, RNA-seq technology was utilized in the analysis of DEGs and novel transcriptome in the spleen of two NE-afflicted chicken lines, lines 6.3 and 7.2 from ADOL that carry the same MHC haplotype (B2) but differ in their response to MD (Briles et al., 1977) or NE infection (Kim et al., 2014).
The draft sequences of spleen were generated from the two genetically disparate chicken lines with approximately 41.7 and 41.6 million sequences reads for NE-afflicted chicken lines 6.3 and 7.2, respectively, in which at least 78% of the reads aligned to the chicken genome (Table 2). In total, the mRNA of 29,997 genes was built with alternatively spliced transcripts. These genes profiles are very attractive, because difference in the expression levels of genes between the two chicken lines may provide clues for underlying protective immune response mechanisms in NE. Recently, several groups were used RNA-Seq to analysis of gene profiles and they show that the minimum 70% of reads were aligned to reference genome and 40% reads were mapped uniquely to the reference genome (Park et al., 2014).
The DEGs profiles of the spleen of two NE-afflicted chicken lines were generated herein, and significantly higher expression was observed in the NE-afflicted line 6.3 than line 7.2, as shown in Table 2. To better understand the gene expression patterns of two NE-afflicted chicken lines, a rigorous algorithm was developed to identify the DEGs between the two lines. By comparing NE-afflicted chicken lines 6.3 and 7.2, more significant DEGs were identified. A total of 2,234 genes were found: 1,239 upregulated and 995 downregulated. DEGs analysis by RNA-seq carried out on hematopoietic stem cells detected a higher number of DEGs than reported previously, which may have been related to the more comprehensive transcriptome discovery method (Park et al., 2014). Herein, GO functional enrichment of the gene expression was performed. Blast2GO software (v.2.7.1) with the GO database was used for the analysis of DEGs in both chicken lines, revealing 135 associated GO biological processes, 88 molecular functions and 22 cellular components in the NE-afflicted chicken line 6.3, and 70 associated biological processes, 10 molecular functions and 54 cellular components in the NE-afflicted chicken line 7.2. When comparing the DEGs between the two NE-afflicted chicken lines, the GO database indicated the annotations to be associated with 141 GO biological processes, 76 molecular functions and 34 cellular components. Most of the DEGs were involved in various immunological responses of the KEGG pathways, such as the JAK-STAT signalling pathway, TGF-╬▓ signaling pathway, toll like receptor signaling pathway, MAPK signaling pathway, and the cytokine-cytokine receptor interaction pathway, particularly in NE-afflicted chicken line 6.3 (data not shown). Changes in the expression of genes in these pathways have also been discovered in the spleen after Clostridium infection, including signal transducer and activator of transcription, growth factor receptor-bound protein, cytokines and cytokine receptor genes (Zhou et al., 2009). Analyzed DEGs in the spleen of broiler chickens after vaccination and challenge by Escherichia coli, finding that most of the DEGs influencing the KEGG pathways were involved in the JAK-STAT signalling pathway, toll like receptor signalling pathway and the cytokine-cytokine receptor interaction pathway. The GO terms in which DEGs of the two NE-afflicted chicken lines were found uniquely helped us to provide insight into identifying the expression of which genes are different, as well as the overall processes defined by those genes. Changes in the genes in these pathways highlight the importance of proper signalling cascades to fight NE infection. The results from DEGs add greater depth to the knowledge base about host response to NE disease.
One of the most important findings from the RNA sequences is the finding of a large number of novel genes in the two NE-afflicted chicken lines. TopHat algorithm mapping to the chicken reference genome was used to find candidates novel genes, and 51% of the consensus sequences generated from the de novo assembly were annotated. This analysis included a large number of transcripts (15,518 contigs) not present in public databases. Overall, chicken line 6.3 showed a substantially larger number of novel genes than line 7.2. The false positive rate needs further investigation. The functional classification of the novel transcripts identified many transcripts specifically involved in the immune response, such as in the MAPK signaling pathway, JAK-STAT signaling pathway, TGF-╬▓ signaling pathway, and toll like receptor signaling pathway. Therefore, a large number of the candidate novel genes were highly diverse, with a large proportion involved in transport, developmental processes, stress response, and cell adhesion. Based on the large number of new exons observed, analyses of the total RNA extracted from the spleen of the two NE-afflicted chicken lines may be a useful approach for the mining of candidate novel genes (data not shown).
In this study, 150 chicken cytokine genes were found to be related to the NE-afflicted chicken lines. Overall, this number is similar to the number of genes identified in duck (150 genes) and zebra finches (150 genes) and is substantially lower than number of mammalian cytokine genes, such as the 230 genes identified in humans and 218 genes in mice (Huang et al., 2013). It is suggested that the RNA-seq analysis performed in this study will provide useful information on the altered expression of innate immune genes, or the discovery of novel genes in the spleen, after NE infection in the two chicken lines. Cytokines, including the IL-2, IL-4, IL-5, TGF-╬▓, and IL-10 family, play a major role in the adaptive immune system (Wright et al., 2013). First, TGF-╬▓ is produced by T cells and many other cell types. It is primarily an inhibitory cytokine, and inhibits the proliferation of T cells as well as the activation of macrophages. It also acts on polymorphonuclear neutrophil granulocytes and endothelial cells to block the effects of pro-inflammatory cytokines (Gandrillon et al., 1999). Among the TGF-╬▓ family, TGF-╬▓1 was significantly increased by 7.34- and 7.46-fold in chicken lines 6.3 and 7.2 after co-infection with EM/CP. In addition, TGF-╬▓ receptors (TGF-╬▓R2, TGF-╬▓RAP1, and TGF-╬▓R1) were also upregulated, and shown to have higher expression in chicken line 6.3 (by 2.18-, 1.22, 3.91-fold, respectively) than in line 7.2 (0.96-, 0.4-, and 3.41-fold, respectively) with p<0.05 (Table S4, Figure 5). Second, interleukin 2 (IL-2) and interleukin 4 (IL-4) are produced by T helper cells, although they can also be produced by cytotoxic T cells to a lesser extent. They are major growth factors for T cells, and also promote the growth of B cells while having the ability to activate NK cells and monocytes (Hemmerle and Neri, 2013). The data obtained herein indicated marked upregulation of IL-2 receptors (IL-2RB and IL-2RG) in the two NE-afflicted chicken lines: by 2.20- and 3.56-fold in chicken line 6.3 and 1.54- and 3.58-fold in line 7.2 for the two genes, respectively. Interleukin 4 receptors were also significantly upregulated in both chicken lines (Supplementary Table S4, Figures 4 and 5).
Interleukin 5 (IL-5) is produced by Th2 cells and functions to promote the growth and differentiation of B cells and eosinophils, as well as activate mature eosinophiles (Johansson et al., 2013). In this study, IL-5 was markedly downregulated in line 7.2 by 0.04-fold, but significantly upregulated by 0.4-fold in line 6.3. Moreover, the expression of IL-5R was significantly increased by 1.22 and 7.26-fold in both lines, respectively. Third, the IL-10 family of cytokines consists of nine members: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, and the more distantly related IL-28A, IL-28B, and IL-29, in mammals. Evolutionarily, IL-10 family cytokines emerged before the adaptive immune response (Wolk et al., 2002). These cytokines elicit diverse host defense mechanisms, especially from epithelial cells, during various infections. IL-10 family cytokines are essential for maintaining the integrity and homeostasis of tissue epithelial layers (Wolk et al., 2002). Herein, 5 genes and 5 receptors of the IL-10 family were observed: IL-10, IL-19, IL-22, IL-26, and IL-28B genes, and IL-20RA, IL-20RB, IL-20RA1, IL-20RA2, and IL-28RA receptors. Among the IL-10 family transcripts, three interleukins (IL-19, IL-22 and IL-26) were downregulated by 2.87- to- 7.84 fold in the two chicken lines with p<0.01 and fold change Ōēź2 (Supplementary Table S4, Figures 4 and 5). The IL-28B gene (IFN-╬╗3) was upregulated in chicken line 6.3 by 2.98-fold but downregulated in line 7.2 by 0.3-fold, while IL-10 was significantly upregulated in both chicken lines by 0.5- and 2.32-fold in lines 6.3 and 7.2, respectively. In addition, the interleukin 10 family receptors, IL-22RA1 and RA2, were downregulated in both chicken lines by 2.11- to 10.08-fold. The IL-20 receptor genes, IL-20RA and IL-20RB, were also upregulated in the two NE-afflicted chicken lines by 1.53- to 3.59-fold (Supplementary Table S4, Figures 4 and 5). Previous studies reported that IL-10 and its family members share common receptors (Commins et al., 2008). Often, however, cytokines have distinct, if not antagonistic, functions. As an example, the signals of both IL-10 and IL-22 go through IL-10R2 to activate JAK1 and TYK2, respectively, thus resulting in STAT3 activation. IL-22, however, also induces serine phosphorylation of STAT3 (while IL-10 does not), an event that is associated with MAP kinase pathway activation (Sabat, 2010).
Of this family of pattern recognition receptors, the TLRs appear to be the most important, and have been the subject of intensive research over the past decade. TLRs are preferentially expressed on immune cells, including macrophages, DCs, monocytes, neutrophils, eosinophils, natural killer cells, platelets, and T and B lymphocytes (Shi et al., 2007). More recently, increasing evidence has indicated that the engagement of TLRs can promote cancer cell growth, induce evasion of immune surveillance, and enhance tumor metastasis and chemoresistance, or rather, induction of tumor cell apoptosis depending on ligands (Gonzalez-Reyes et al., 2010). The avian genome encodes 10 functional TLRs that are located either on the cell surface or within endosomes (Ramasamy et al., 2014). In a previous study, the TLR genes were found to be differentially expressed during the early stages of infection by Salmonella Pullorum in chicks, with significant upregulation of the expression of TLR2, TLR4, and TLR6 in the gastrointestinal tissues, but significant downregulation of TLR3 and TLR15 (Ramasamy et al., 2014). In addition, the expression of TLR3 and TLR7 was significantly upregulated in both susceptible and resistant chicken lines treated with polyionosinic-polycytidylic acid, being significantly higher in the resistant chickens compared with susceptible chickens (Haunshi and Cheng, 2014). In this study, the expression of 6 TLR genes (TLR1, TLR5, TLR6, TLR7, TLR15, and TLR21) was detected. Expression of the 6 TLR genes was significantly upregulated by 2.28- to 8.04-fold in chicken line 6.3, while that of 5 of the TLR genes (excluding TLR5) was significantly upregulated by 2.23- to 8.26-fold in chicken line 7.2 at p<0.01 (Supplementary Table S4, Figures 4 and 5). These results revealed that genes of the TLR pathway play an important role in the pathogenicity of EM/CP co-infection model of NE. The findings are helpful for understanding the molecular basis of pathogenesis and the underlying mechanism of NE response, and also provide strong evidence of TLR involvement in the innate immune response to NE disease in poultry.
We also investigated 139 immune related genes for which higher expression was observed among the chicken lines (18 genes upregulated in line 6.3 and 12 genes in line 7.2, comparatively with p<0.01 and fold Ōēź2). In a previous report, search of the 450,000 sequences in the chicken expressed sequence tag collection enabled identification of 185 immune-related sequences which are also members of the cytokines, chemokines, antigens, cell surface proteins, receptors and MHC-associated genes (Smith et al., 2004). In the present study, most of the immune-related genes were also members of the antigens, cell surface proteins, receptors and MHC-associated genes, STAT family, TRAF family, interleukins and differentiation antigens. An interesting finding is that most of the immunoglobulin (Ig) genes were upregulated in the two NE-afflicted chicken lines. However, some of the Ig genes were more highly expressed in the MD-resistant chicken line 6.3 than in the spleen of the susceptible chicken line 7.2, including IgJ, IgA, Ig rearranged H-chain, Immunoglobulin heavy chain variable region, and Ig germline heavy chain VD region (Supplementary Table S3). The investigation of these genes will advance basic avian immunology of immunoglobulins and will pave the way for large-scale immune-related microarray experiments, providing new insight into functional and evolutionary studies.
Moreover, the CD molecular genes were also markedly upregulated: 24 CD genes in chicken line 6.3 and 23 CD genes in line 7.2 with p<0.01 and fold Ōēź2. The role of CD presented on the cell membrane is to allow molecular response to pathogens. CD molecular genes remain specific to a development period or as characteristic markers until destruction of the cell membrane. The majority of CD antigens are involved in immune functions of organisms and include the receptors for antigens, MHC glycoproteins, adhesive molecules, receptors for immunoglobulins, receptor for complement, receptors for lymphocytes and other growth and differentiation factors, membrane enzymes or transport molecules and other molecules (Fabryova and Simon, 2009).
Additional validation of the RNA-seq analysis was performed through qRT-PCR analysis of selected genes in the two NE-afflicted chicken lines. Gene expression changes for the 14 genes as observed by qRT-PCR were compared for the two chicken lines, as shown in Figures 7 and S1. Of those analyzed, the gene expression levels of 7 genes (IL-16, LILRB3, CXCL13, IL-7R, LILRB5, TLR21, and TNFAIP2) were higher in chicken line 6.3 than in line 7.2, while the other 7 genes (CCL3, IL-34, LILRB1, LILRB4, CCL4, CSF3R, and IL-1R2) showed significantly higher expression in chicken line 7.2 (Figures 7 and Supplementary Figure S1). All altered genes examined herein showed similar responses to co-infected EM/CP exposure in the qRT-PCR and RNA-Seq analyses. A high correlation between RNA-seq and qRT-PCR was observed at the range of 0.81 to 0.87 (p<0.01) as shown in Figure 8. Quantitative RT-PCR validation was also an important indication that the pooled samples (2 pools of each 10 chickens) used for RNA-seq analysis reflected the expression levels in the individual pools. While pooled samples obviously could have masked individual variation, the goal in the present study was to gain a broader understanding of gene responses to NE infection in the spleen and to provide insights into important pathways and processes.
We draw four noteworthy conclusions from our results. First, using next-generation sequencing technology, we generated the first draft sequence for two chicken lines, one of which is a natural host of NE disease. Second, we identified 139 immune-related genes that were differentially expressed in the two NE-induced chicken lines. Further efforts identified 150 cytokines with differential expression in the two chicken lines. Third, we performed a deep transcriptome analysis to characterize gene expression profiles and to identify genes that are responsive to NE disease. Fourth, we found 15,518 candidate novel genes that may be involved in the host immune response to NE disease whose infection was examined in the two chicken lines. Overall, the genes of chicken line 6.3 were more highly expressed than those of chicken line 7.2 in response to NE induction. This dataset will be helpful for gene discovery, function, mapping, and genomic evaluation in chickens. Moreover, the significant DEGs will useful for future studies to understand the regulation and function of signalling pathways in the two genetic chicken lines of the present study. Collectively, the results generated in this study have provided information that our current knowledge of how chicken genes control NE disease and further study to develop disease resistance markers for molecular breeding.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







