Biocomputational Characterization and Evolutionary Analysis of Bubaline Dicer1 Enzyme
Article information
Abstract
Dicer, an ribonuclease type III type endonuclease, is the key enzyme involved in biogenesis of microRNAs (miRNAs) and small interfering RNAs (siRNAs), and thus plays a critical role in RNA interference through post transcriptional regulation of gene expression. This enzyme has not been well studied in the Indian water buffalo, an important species known for disease resistance and high milk production. In this study, the primary coding sequence (5,778 bp) of bubaline dicer (GenBank: AB969677.1) was determined and the bubaline Dicer1 biocomputationally characterized to determine the phylogenetic signature among higher eukaryotes. The evolutionary tree revealed that all the transcript variants of Dicer1 belonging to a specific species were within the same node and the sequences belonging to primates, rodents and lagomorphs, avians and reptiles formed independent clusters. The bubaline dicer1 is closely related to that of cattle and other ruminants and significantly divergent from dicer of lower species such as tapeworm, sea urchin and fruit fly. Evolutionary divergence analysis conducted using MEGA6 software indicated that dicer has undergone purifying selection over the time. Seventeen divergent sequences, representing each of the families/taxa were selected to study the specific regions of positive vis-à-vis negative selection using different models like single likelihood ancestor counting, fixed effects likelihood, and random effects likelihood of Datamonkey server. Comparative analysis of the domain structure revealed that Dicer1 is conserved across mammalian species while variation both in terms of length of Dicer enzyme and presence or absence of domain is evident in the lower organisms.
INTRODUCTION
Dicer belongs to family of ribonuclease type III (RNase III) enzyme that plays an important role in the biosynthesis of miRNA by cleaving the pre-miRNA into mature double-stranded miRNA (ds-miRNA) of ~21 nucleotide length. Structurally, Dicer1 has mainly two RNase III domains along with helicase and Piwi/Argonaute/Zwille (PAZ) domains. However, the complexity of the domains varies among divergent organisms. Dicer protein is present in most of the eukaryotes with the exception of Saccharomyces cerevisiae (brewer’s yeast). Single copy of Dicer gene occurs in nematodes (Caenorhabditis sp.), mammals and poikilothermic vertebrates. Fungi (e.g. Neurospora crassa), insects (e.g., Drosophila and mosquito; Anopheles gambiae and Aedes aegypti), and possibly all arthropods encode two Dicer genes, Dicer-1 (Dcr1) and Dicer-2 (Dcr2), which are involved in processing pre-miRNA for association with RNA-induced silencing complex (RISC) and siRNA production in the RNAi pathway, respectively (Lee et al., 2004). Plants (e.g. Arabidopsis thaliana, poplar, rice etc.) encode four Dicer homologues (Dcl-1 to 4), each having specialized functions. Dcl-1 (processes mature miRNA), Dcl-3 (generates siRNA), and Dcl-4 (trans-acting siRNA biogenesis), each contains two double stranded RNA binding domains (dsRBDs), while Dcl-2 (produces siRNA against viral infection) contains only one dsRBD (Kurihara and Watanabe, 2004; Gasciolli et al., 2005; Xie et al., 2005).
Although Dicer enzyme have been well characterized in human and plants but reports on bovine Dicer1 sequence and expression as well as its evolutionary studies are in vogue. Indian water buffalo (Bubalus bubalis) is an important species for milk production and as a mammalian model organism for comparative genomics and biological studies. Besides, no report is available on biocomputational analysis of bubaline Dicer1 coding sequence with regard to its evolutionary perspectives. Therefore, the present work was designed to determine the primary cDNA sequence of bubaline Dicer1 with an aim to study the evolution of bubaline Dicer1 coding sequence.
MATERIALS AND METHODS
Collection of blood samples
Peripheral blood was aseptically collected in anticoagulant from the jugular vein of adult, healthy, male, Murrah buffalo maintained at Dairy farm, Guru Angad Dev Veterinary and Animal Sciences University, Ludhiana, India. The work was approved by the Institutional Animal Ethics Committee (IAEC) and all the protocols followed were as per the guidelines of the committee.
cDNA amplification and custom sequencing
Primers (Table 1) targeting taurine Dicer1 coding sequence (Genbank Acc. No. NM_203359) were designed to amplify overlapping fragments of partial coding sequence (cds) of bubaline dicer using the online tool Primer3 (Untergrasser et al., 2012) and quality-checked by IDT Oligoanalyzer 3.1 (http://eu.idtdna.com/analyzer/applications/oligoanalyzer/).

Detail of primer-pairs (sequence, annealing temperature, amplicon length and GC %) used for amplifying overlapping fragments of Dicer1 cds
Total RNA extraction and cDNA synthesis
Total RNA was isolated using TriZol (Ambion) from the leukocytes following red blood cells lysis. The quantity and purity of RNA was checked spectrophotometrically using NanoDrop 1000 (Thermo Scientific) and the RNA templates having absorbance ratio (260/280) between 2.0 and 2.1, were subjected to first strand cDNA synthesis, using RevertAid First Strand cDNA synthesis kit (Thermo Scientific, Vilnius, Lithuania), according to the manufacturer’s instruction.
cDNA amplification and cloning
Polymerase chain reaction (PCR) was then conducted in 25 μL reaction volume (with final concentration of components: 1X reaction buffer, 15 mM MgCl2, 0.4 μM each of primer pair and 1 unit of Taq polymerase recombinant) using the cDNA (2 μL) as template in Veriti (Applied Biosystems, ThermoFisher Scientific Brand, Waltham, MA, USA) thermocycler. The conditions of PCR amplification was: initial denaturation (T = 95.0°C, 3 min); followed by 35 cycles of denaturation (T = 94.0°C, 30 s); annealing (detail in Table 1, 30 s); extension (T = 72.0°C, 0:45 min); and final extension (T = 72.0°C, 5 min). Amplified products were subjected to agarose gel electrophoresis and visualized using Chemidoc XRS Gel documentation system (Biorad, Hercules, CA, USA).
Purified PCR Products for each fragment were ligated in pGEMT-easy vector (Promega, Madison, Fitchburg, WI, USA) cloning vector and transformed into competent DH5α strain of E. coli. The transformed cells were plated on Luria-Bertani agar plate containing ampicillin (50.0 μg/mL). Recombinant colonies were subjected to plasmid isolation using the GeneJET Plasmid Miniprep Kit and confirmed by restriction endonuclease digestion, using EcoRI for the release of insert. The plasmids were then custom sequenced in both directions at DNA Sequencing Facility, Department of Biochemistry, University of Delhi, South Campus, New Delhi, India.
Sequence analysis
Sequence trimming and submission
The forward and reverse sequences of the plasmids were screened for removal of non-essential vector sequences using BLASTn (Altschul et al., 1990) and vecScreen (http://www.ncbi.nlm.nih.gov/tools/vecscreen/) online tools. The individual partial sequences were then submitted to NCBI, Nucleotide databank. The partial cds sequences were combined to get the complete Dicer1 coding sequence.
Downloading homologous sequences
The final Dicer1 complete coding sequence was subjected to BLASTn (Altschul et al., 1990) to retrieve homologous cds of Dicer belonging to divergent species available at the NCBI database (http://blast.ncbi.nlm.nih.gov/) based on higher percent identity and E-value (<10−5). A total of 115 Dicer cds and their respective amino-acid sequences from divergent species were downloaded and saved in FASTA format.
Multiple sequence alignment
The amino acid sequences of divergent species were subjected to multiple sequence alignment using DNA Star (Lasergene, DNASTAR. Inc., Madison, WI, USA) and MAFFT online software (http://mafft.cbrc.jp/alignment/software/index.html) to identify the evolutionarily conserved regions of Dicer1 Ribonuclease III enzyme among animals. Similarly, ruminant specific Dicer1 amino acid sequences were aligned in order to determine the extent of evolutionary conservedness of the enzyme. Finally, seventeen divergent sequences, representing each of the families/taxa were selected to study the specific regions of positive vis-à-vis negative selection.
Phylogenetic inference
The MEGA6 software (Tamura et al., 2013) was used for determining the best evolutionary model, for phylogenetic tree construction, estimation of evolutionary divergence, determining the amino acid composition and estimation of selection pressure on coding sequences. The best evolutionary model was determined based on the least Bayesian Information Criterion (BIC) scores. The Akaike Information Criterion, corrected and maximum likelihood values were determined for each of the models. The phylogenetic tree of these sequences was inferred using maximum likelihood method (with 500 bootstrap replication) using the selected best model, with 5 discrete Gamma categories for rates among sites, with complete deletion of the missing data. Phylogenetic trees were constructed for both the sets of data (115 as well as 17 selected amino acid sequences).
Estimation of evolutionary divergence between species
The evolutionary divergence between all ruminant amino acid sequences, and the 17 sequences representing specific divergent families were estimated to obtain the base substitution per site using the Jones-Taylor-Thornton (JTT) matrix-model (Jones et al., 1992) with Gamma parameter 5 and 500 bootstrap replicates. The evolutionary divergence values (substitution per site) were graphically represented by the use of Heatmap, using WGCNA package of R program (Version 3.0.2, Los Angeles, CA, USA).
Estimation of selection pressure on coding sequences
The nucleotide sequences of Dicer coding sequences of divergent species were subjected to codon based tests, in order to determine the effect of evolutionary forces that have tailored its encoded products. The numbers of synonymous (dS) and nonsynonymous substitutions (dN) per synonymous and non-synonymous sites, respectively, were used to calculate the test statistic (dN-dS) along with the probability of rejecting the null hypothesis that the codons have evolved through neutral selection (dN = dS), against the alternative hypothesis of evolution of the codon through positive selection (test of positive selection; dN>dS) or through purifying selection (test of purifying selection; dN<dS).
Analyzing the positive and negative sites
The estimation of selection pressure, based on the rate of synonymous (dS) and non- synonymous (dN) mutations, on different codons of Dicer enzyme belonging to seventeen divergent coding sequences representing each of the families/taxa, was done using Datamonkey online server (http://www.datamonkey.org./). Different models were checked for the study, namely, single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL) and random effects likelihood (REL). Finally, the REL model was considered for interpretation of results. Branch-site REL analysis for estimating the episodic diversifying selection among the divergent species was carried out.
Domain architecture of dicer
The various domains present in the Dicer enzyme of the divergent 17 sequences were identified using BlastP to find out the variations in length of different motifs. The comparative domain architecture was graphically represented.
Amino acid composition
The frequency of the amino acids was calculated in each of the 17 divergent sequences and graphically represented as heatmap. Two-tailed paired t-Test, assuming equal variance, was conducted between the bubaline dicer amino acid composition and other 16 species, using Systat software (Systat Inc., San Jose, CA, USA).
RESULTS AND DISCUSSION
Cloning of bubaline Dicer coding sequence
The various partial overlapping fragments of bubaline dicer1 enzyme were cloned using pGEMT-easy vector and recombinant plasmids were confirmed by restriction endonuclease digestion, using EcoRI for the release of insert (Figure 1, RE digestion of clones DR2, DR3, DR5, DR6, RN5, RN6, and RSE2). The positive recombinant clones were further sequenced; and the individual partial sequences were submitted to the nucleotide database at NCBI or DDBJ (GenBank Acc. No.: KF724684.1, AB909393.1, AB889485.1, AB909391.1, KF724685.1, AB889486.1, AB924056.1, AB909392.1, AB906337.1, KF056324.1, and KF021228.1). The final complete bubaline-Dicer1 cds sequence was interpreted by sequence alignment and submitted to the DDBJ (AB969677.1). The coding amino acid sequence of Bubaline Dicer1 enzyme is shown in Figure 2.

Confirmation of clones by EcoRI RE digestion for release of insert, run on 1.5% agarose gel. (A) Lane 1: Insert release of ~913 bp (DR2); (B) Lane 1: Insert release of ~518 bp (RN5); (C) Lane 1: Insert release of ~928 bp (DR3), Lane 2: Insert release of ~910 bp (DR5), Lane 3: Insert release of ~927 bp (DR6); (D) Lane 1: Insert release of ~789 bp (RSE2); (E) Lane 1: Insert release of ~1,009 bp (RN6). EcoRI restriction endonuclease (RE) enzyme isolated form strain of E. coli. M: 1 Kb plus DNA ladder.

Coding amino acid sequence of the Bubaline Dicer1 enzyme.
Sequence analysis
The best model i.e. JTT+Gamma (G) was selected for further evolutionary analysis of the 115 amino acid sequences, based on the lowest BIC score of 31,275.44. The model with the lowest BIC score can best represent the substitution pattern in the coding sequences (Nei and Kumar, 2000). The Gamma distribution (+G) adjusts the non-uniformity of the evolutionary rates among the sites of the codons (Tamura et al., 2013). The best model selected for analyzing the 17 divergent amino acid sequences was Le and Gascuel (LG)+G (Le and Gascuel, 2008), based on the lowest Bayesian Index Score (44,127.7841).
Phylogenetic inference
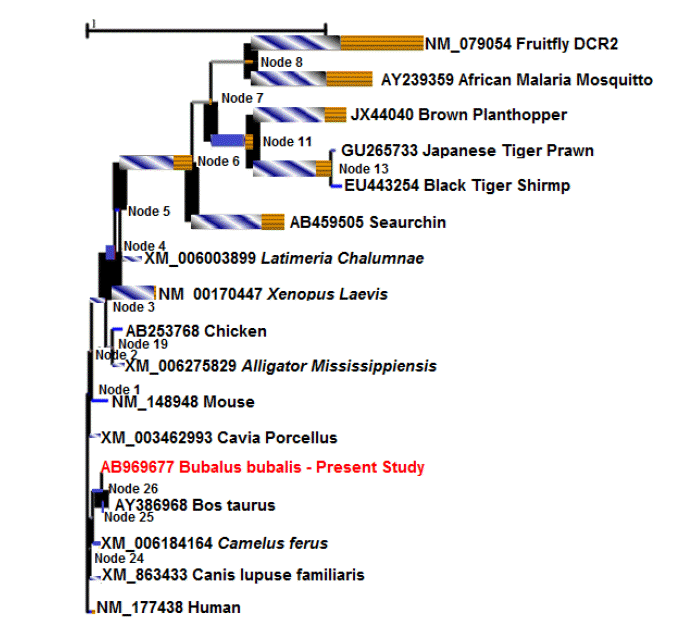
The phylogenetic tree was constructed subjecting 115 Dicer amino acid sequences to maximum likelihood with 500 bootstrap resampling (MEGA 6) (Figure 3). The evolutionary tree demonstrated that all the transcript variants of Dicer1 belonging to a specific species were coming within the same node. It was observed that sequences belonging to same family or order were forming a cluster; therefore, these sequences were merged and represented by same leaf (terminal OTU) for better resolution of the phylogeny. The bubaline Dicer1 sequence (including the transcript variants) formed one clad with that of cattle and yak (bootstrap value 92). Higher bootstrap value (100) is observed among the ruminants (viz. buffaloes, cattle, yak and sheep, goat, antelope), indicating higher consistency of the given data for taxonomical bipartitioning (Hedges, 1992). Bootstrap values do not indicate how accurate the tree is, however, it indicates the stability of the branching pattern. In the present study, the higher bootstrap values of the branches of avian with reptiles (bootstrap value 96); and the mammals, avians, reptiles with that of other species like, Sarcophilus harrisii (Tasmanian devil, a carnivorous marsupial ), Xenopus laevis (African clawed frog), Western clawed frog, Latimeria chalumnae (West Indian Ocean coelacanth), Ctenopharyn godonidella (Grass carp), zebra fish, Hymenolepis microstoma (Rodent tapeworm)) (bootstrap value 98) clearly signify the stability of branching pattern.

Phylogenetic tree of Dicer1 enzyme among the animal species, constructed using maximum likelihood method (500 bootstrap resampling). The species belonging to same family that were forming a cluster have been merged as a single operational taxonomic unit (OTU). The bootstrap value (>50) have been indicated along the nodes.
Another phylogenetic tree was constructed using the seventeen divergent sequences, each representing a class/order, for comprehensible interpretation (Figure 4) of the previous result. The phylogenetic tree depicted that the Bubaline Dicer1 is closely related to that of cattle. The mammalian dicer sequence were clustering together and separate from the other lower organism. The prawn and shrimp formed a clad separate from that of insects (mosquito and plant hopper). Fruit fly Dicer2 is the most distantly related from all the species as depicted by a separate node. The tree indicated that Dicer1 enzyme have undergone natural selection through the time in accordance with the requirement of environment.

Phylogenetic tree constructed from 17 divergent Dicer amino acid sequences, using maximum likelihood method (500 bootstrap resampling). The tree is drawn to the scale and branch length (number of substitutions per site) as well as bootstrap value (>50) have been indicated in the tree.
Report suggests that D. melanogaster Dcr2 and Ago2 are among the fastest evolving genes in this organism, perhaps as a result of a co-evolutionary ‘arms race’ with viral pathogens (Obbard et al., 2006). Murphy and coworkers (2008) studied the phylogenetic and evolutionary relationship of the four major proteins (Dicer, Argonaute, RISC RNA-binding proteins, and Exportin-5) involved in miRNA biogenesis and suggested lineage specific expansion of Dicer1 in plants and invertebrates. Similar observations regarding the phylogenetic localization of vertebrate vis-à-vis Dicer were made in the present study. Cerutti and Casas-Mollano (2006) examined the taxonomic distribution and the phylogenetic relationship of key-components of the RNA interference (RNAi) machinery in members of five eukaryotic super-groups. While insect Dcr1 clusters with all other animal Dicers, Dcr2 is much more divergent and forms a paralogous clade. Stowe et al. (2012) determined the primary cds of porcine Dicer (pDicer) and studied its expression in porcine oocytes and early stage embryos as well as its phylogenetic perspective. The pDicer coding sequence was found to be highly conserved with bovine dicer and pDicer being the most conserved to the human Dicer than the mouse homolog. The Droshophila and C. elegans were found to be most distant among all the species.
Estimation of evolutionary divergence between species
The evolutionary divergence estimates among the primates viz. Pan paniscus (Pygmy chimpanzee), Pan troglodytes (Common chimpanzee), Homo sapiens (Human), Gorilla, Pongoabelii (Sumatran orangutan), Nomascus leucogenys (Northern white-cheeked gibbon) varied between 0.000 to 0.003, while the highest amount of divergence among the mammalian species was 0.006 between Callithrix jacchus (Common marmoset/New World monkey) and Pygmy chimpanzee. Interestingly, the Dicer1 sequence of the African Elephant is found close to that of the primates. Sequence divergence is evident among the evolutionarily distant species like Sarcophilus harrisii (Tasmanian devil), Xenopus laevis (African clawed frog), Western clawed frog, Latimeria chalumnae (West Indian Ocean coelacanth), Ctenopharyn godonidella (Grass carp), zebra fish, Hymenolepis microstoma (Rodent tapeworm).
The results indicated that the number of amino acid substitutions per site was minimum for pair of sequences belonging to same clad, as it is evident from the phylogenetic tree. Hymenolepsis microstoma (tapeworm) has shown the highest amount of evolutionary divergence with the several taxa. While the Dicer1 sequence of the primates have revealed the least amount of divergence among themselves. Interestingly, the divergence of various Dicer1 transcript variants was negligible within a species. It indicates that the variants have evolved from the same ancestral sequence no selection pressure has favored any particular variants.
The evolutionary divergence among the seventeen divergent animal species has been represented as heat map (Figure 5). The least rate of evolutionary distance (0.009) was observed between bubaline and cattle dicer enzyme while highest rate evolutionary distance (>2.3) was observed between fruitfly and rest of the species. The heat map color bar ranged from white-black, indicating lowest to highest rate of evolutionary divergence. It was observed that evolutionary divergence was lower (indicated by white color) among all mammalian species and also between prawn and shrimp, while species belonging to lower class showed intermediatary divergence (grey color) with other species. Maximum divergence (dark grey-blak color) was observed between fruitfly and other species. Among the ruminant species, evolutionary divergence of Dicer1 enzyme is very less as evident by the maximum value of 0.022 for camel (i.e. a pseudo-ruminant) and cattle. The buffalo transcript variants; goat and Tibetan antelope; show no divergence (value 0) among themselves and represented by white color. Moderate divergence is visible among camel and other species (dark grey color) (Figure 6).
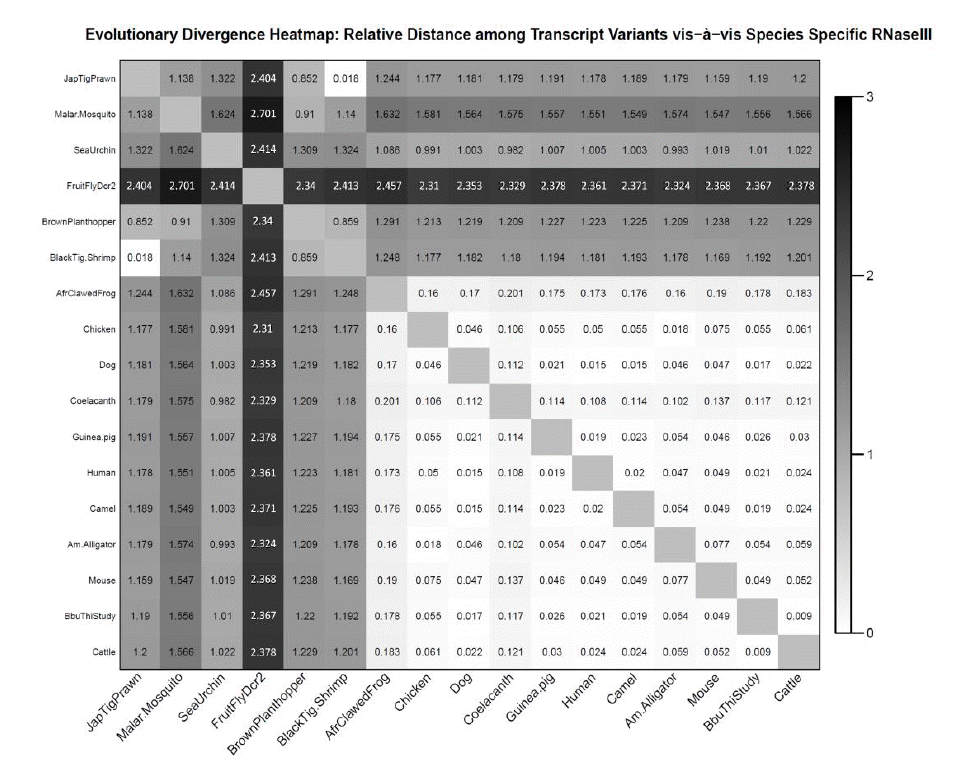
Evolutionary divergence heat map: realtive distance among seventeen divergent animal species based on Dicer sequence.

Evolutionary divergence heat map:realtive distance among ruminants species based on Dicer1 sequence.
Mukherjee et al. (2012) studied the evolution of Dicer in eukaryotes (animals and plants) and showed that Dicer genes duplicated and diversified independently in early animal and plant evolution, coincident with the origins of multi-cellularity. Similar result of gene duplication and evolution of Dicer for various functions was presented by Gao et al. (2014), in case of invertebrate species.
Estimate of selection pressure for various codons
The value of test statistic (dN-dS) determines whether the codon has undergone positive, negative or neutral selection; where dN and dS represents rates of synonymous (S) substitution per synonymous site and non- synonymous (N) substitution per non-synonymous site, respectively. The analysis for codon-based test of purifying selection for analysis between all the divergent sequences indicated that the null hypothesis of strict neutrality (i.e. the numbers of synonymous substitutions per site (dS) commensurate the number of non- synonymous (dN) substitutions per site) of Dicer1 sequence, has been rejected (Table 2). It clearly indicates that the Dicer1 has experienced purifying selective pressure during evolution. The transcript variants have not been selected against as the null hypothesis of neutral selection (i.e. dN = dS) is not found significant for many of the transcript variants within a same species.

Codon-based test for analysis of purifying selection (dN<dS) between Bubaline Dicer1 sequence and that of other divergent species
Results of various studies (Kimura, 1983; Nei, 1987; Li, 1997) suggest that rate of amino acid substitution is determined by the stringency of structural and functional constraints and hence proteins having very stringent structural and/or functional requirements, is subject to a strong negative selection pressure limiting the number of changes in the gene product.
Analyzing the positive and negative sites
Datamonkey results for 17 representative divergent sequences: Different models were used for the study namely SLAC, FEL, and REL. The SLAC being the most conservative model giving minimum number of false positive and false negative result revealed 84 positively and 140 negatively selected sites. The graph plot of SLAC is presented in Figure 7. The graph plot is a diagrammatic representation of dN-dS values versus the codons. The FEL model showed 312 positively and 359 negatively selected sites. The REL model which is the least conservative of the models selected for the study and it revealed 161 positively and 127 negatively selected sites.
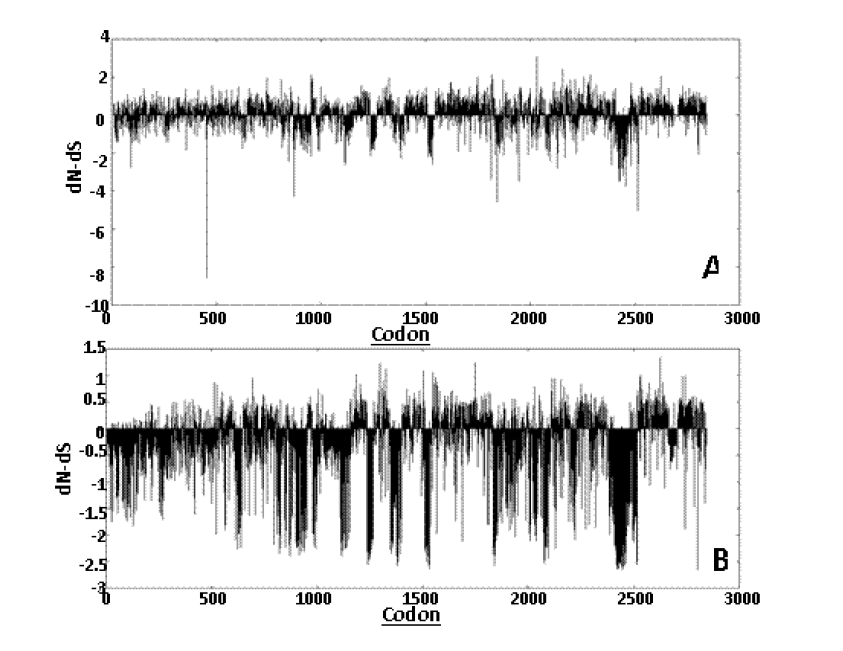
Graphical representation of the dN-dS test statistic versus the codon positions obtained from SLAC (A) and REL (B) analyses. SLAC, single likelihood ancestor counting; REL, random effects likelihood.
Branch site REL analysis results reveled 10 branches under episodic diversifying selection at p≤0.05. Branch-site tests measure selective pressure by estimating omega (ω) value i.e. the ratio of non-synonymous (b) to synonymous (a) substitution rates, and if a proportion of sites in the sequence provides statistically significant support for ω>1 along the lineages of interest, then episodic positive selection is inferred (Pond et al., 2011). The phylogenetic tree scaled on the expected number of substitutions/nucleotide is given in the Figure 8. The strength of selection has been indicated by different bar/line, bar with horizontal lines corresponds to ω>5, blue bar/line to ω = 0 and bar with diagonal lines to to ω = 1. The width of each bar component represents the proportion of sites in the corresponding class. Thicker branches have been classified as undergoing episodic diversifying selection by the sequential likelihood ratio test at corrected p≤0.05.
Phylogenetic tree of seventeen divergent animal species based on Branch site REL result depicting the episodic diversifying selection. REL, random effects likelihood.
Previous studies have found that antiviral Dicer2 is under intense positive selection in Drosophila melanogaster and across the Drosophila phylogeny (Obbard et al., 2006; Heger and Ponting, 2007; Kolaczkowski et al., 2011). A study conducted by Mukherjee et al. (2012) established that Dicer2 DEAD (Aspartate-Glutamate-Alanine-Aspartate) box protein/Helicase and PAZ domains have experienced positive selection in flies using branch-sites analyses to identify adaptive protein-coding changes.
Domain architecture of dicer enzyme
The important domains and their location in dicer enzyme among the divergent animal species have been compared (Figure 9). For the graphical representation, the longer domain of DEAD-like helicases superfamily (DEXDc) have been considered among all the species and for rest of the domains the limits mentioned in the flat-file has been considered as the respective domain length ignoring the possible larger length. The two domains RIBOc 2 and Double-stranded RNA binding motif are located within the dsRNA-specific ribonuclease (RNC). The domains i.e. PAZ and two ribonuclease (RIBOc1 and RIBOc2) are present is all the species. The DEXDc domain is present in most of the species expect Japanese tiger prawn and Black tiger shrimp. It is clearly seen that the domain architecture, i.e. length and position of the various domains, is same in mammalian dicer enzyme. Whereas, in lower organisms there is more variation both in terms of size (amino acid count) of dicer enzyme as well as organization of the domains. It may be concluded that dicer enzyme have gradually evolved from lower organism with more variation to a much stable form in mammalian species.

Comparative representation of domains of Dicer enzyme of divergent animal species.
Variation in the domain architecture of Dicer-like proteins in species belonging to five eukaryotic supergroups have been demonstrated by Cerutti and Casas-Mollano (2006), with only two RNase III catalytic motifs of Dicer domains being predominantly conserved as a fusion across the eukaryotic spectrum. Mukherjee et al. (2012) also identified key changes in Dicer domain architecture and sequence leading to specialization in either gene-regulatory or protective functions in animal and plant paralogs. The organization of the functional domains of the Dicer family has been studied by Gao et al. (2014), among the invertebrate organisms. The study found significant variability in domain organization with Taenia solium Dicer2 processing only one RNase III domain; the loss of the DEAD domain in Dicer1 of mollusks, annelids, platyhelminths and most arthropods; and the absence of the PAZ domain in Dicer2 sequences of S. mediterranea (platyhelminth) and T. adhaerens (metazoa) species.
Amino acid composition
The frequency of the amino acids in Dicer enzyme across the 17 divergent sequences has been graphically represented as Heatmap (Figure 10). Glutamine and Leucine showed maximum proportion in Dicer enzyme across all the sequence while tryptophan was present in lowest proportion. The t-test between the bubaline dicer composition and other 16 species revealed no difference (p = 1.00). It suggests that the Dicer enzyme can be considered one of the most conserved enzyme among animals, however, some difference in amino acid composition is evident from the heatmap (Figure 10) of amino acid composition.
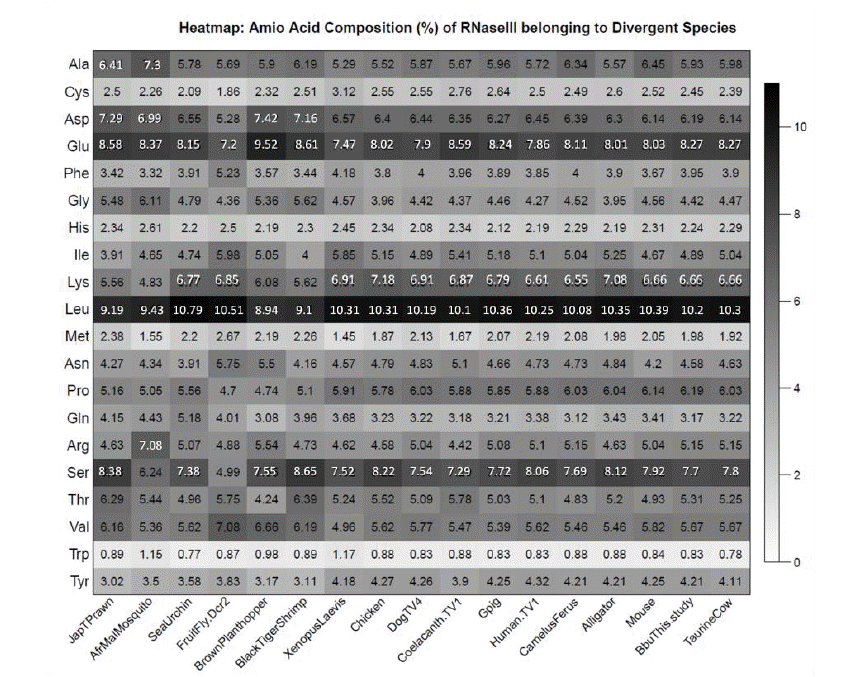
Heatmap of amino acid percentage of Dicer (ribonuclease type III) belonging to divergent species.
Graur (1985) found high correlation between the rate of amino acid substitution of a protein and its amino acid composition, by analysing mammalian genes, and proposed that composition is the main factor in determining the rate of evolution of proteins, whereas functional constraints have only a minor effect. However, Tourasse and Li (2000) showed that rate of protein evolution is only weakly affected by amino acid composition but is mostly determined by the strength of functional requirements or selective constraints. Therefore, regions or individual sites critical for the function within a given protein, such as catalytic sites or binding domains, are generally better conserved than the rest of the molecule. In the present study also the various domains of bubaline Dicer are found to be highly conserved among the divergent animal species.
IMPLICATIONS
Evolutionary studies provided the evidence of purifying selection in the Dicer enzyme among the animal species. The primary coding sequences of bubaline Dicer would further be useful to carryout expression studies in the buffaloes and domains architecture of bubaline Dicer revealed in this study may be used for its biochemical and functional characterization.
ACKNOWLEDGMENTS
The authors thankfully acknowledge the Science and Engineering Research Board (SERB) and Department of Science and Technology (DST), Government of India, for providing the funds (SR/FT/LS-22/2011) to conduct the research work. It is also certified that there is no conflict of interest among the authors.