Bovine Genome-wide Association Study for Genetic Elements to Resist the Infection of Foot-and-mouth Disease in the Field
Article information
Abstract
Foot-and-mouth disease (FMD) is a highly contagious disease affecting cloven-hoofed animals and causes severe economic loss and devastating effect on international trade of animal or animal products. Since FMD outbreaks have recently occurred in some Asian countries, it is important to understand the relationship between diverse immunogenomic structures of host animals and the immunity to foot-and-mouth disease virus (FMDV). We performed genome wide association study based on high-density bovine single nucleotide polymorphism (SNP) chip for identifying FMD resistant loci in Holstein cattle. Among 624532 SNP after quality control, we found that 11 SNPs on 3 chromosomes (chr17, 22, and 15) were significantly associated with the trait at the p.adjust <0.05 after PERMORY test. Most significantly associated SNPs were located on chromosome 17, around the genes Myosin XVIIIB and Seizure related 6 homolog (mouse)-like, which were associated with lung cancer. Based on the known function of the genes nearby the significant SNPs, the FMD resistant animals might have ability to improve their innate immune response to FMDV infection.
INTRODUCTION
Foot-and-mouth disease (FMD) is a highly contagious disease affecting cloven-hoofed animals including cattle, pig, sheep, and goat with the clinical signs of fever, lameness and vesicular lesions on the mouth, tongue, feet, snout, and teats of infected animals. The disease causes severe economic loss and has a devastating effect on international trade of animal or animal products. In the past, FMD outbreaks have occurred in every livestock-containing region of the world, except for New Zealand. Currently, FMD is enzootic in all continents except Australasia and North America which have been free for many years (Grubman and Baxt, 2004). Although systematic vaccination has succeeded in Western Europe with a great reduction of FMD incidence, the presence of live virus in some batches of vaccine and the escape of virus from manufacturing facilities have been responsible for a large proportion of the outbreaks (Bachrach, 1985). In 2010, South Korea had FMD outbreaks which started in the south Eastern part of the Korean peninsula and spread almost nationwide. Because of a national vaccination program and an eradication policy, no further outbreaks of FMD were reported since 2011 in Korea.
The causative agent of FMD is the foot-and-mouth disease virus (FMDV), which is an aphthovirus of the family Picornaviridae (Belsham, 1993). If an animal is infected with FMDV, antibodies against the virus are elicited by the immune system of the host animal. The FMDV contains a positive strand RNA genome which encodes the structural protein (SP) of four subunit proteins (VP4, VP2, VP3, VP1) and non-structural proteins (NSP; L, 2A, 2B, 2C, 3A, 3B, 3C, and 3D) (Sobrino et al., 2001; Grubman and Baxt, 2004). The 60 copies of the structural proteins are components of its icosahedral shape and the non-structural proteins are responsible for the replication of viral genome in the host cells. In the infected animals, antibodies against non-structural proteins are produced. Nowadays, the FMD vaccines made of inactivated and purified FMDV particles nearly exclude the non-structural proteins due to the advanced purification technology. The vaccinated animal could be discriminated from the infected by the absence of the antibodies against non-structural proteins.
The link between diverse immunogenomic structures of cattle and the immunity to FMDV has been investigated in a limited way. Some studies focused on the bovine major histocompatibility complex (MHC) region to find some haplotypes closely related with the immunity elicited after vaccination or the defense of cattle against FMD infection (Haghparast et al., 2000; Gerner et al., 2007) or to find epitopes in FMDV to react with MHC molecules (Guzman et al., 2008). Quantitative trait loci (QTL) for variation in immune response to a FMDV peptide were identified in Holstein cattle and the majority of QTL were found outside MHC locus (Leach et al., 2010). Since a high-density bovine chip based on the whole genome sequence is available, a genome-wide association study (GWAS) is a more realistic approach to understanding the mechanism of host resistance to FMDV. In this study, we performed genome wide association study to identify loci associated with FMDV resistance in Holstein using high density bovine single nucleotide polymorphism (SNP) chip.
MATERIALS AND METHODS
Foot-and-mouth disease virus diagnosis and animal sampling
In order to perform GWAS study for identifying FMDV resistant loci in cattle, we collected a total of 96 female Holstein (26 infected and 70 resistant animals) reared in four field dairy farms which have been affected by FMD and some of the animals were found to be infected (Table 1). The distinction between the FMD susceptible or resistant animals was determined twice using 2 different brands of enzyme-linked immunosorbent assay kits (JENOBIOTECH, Median Diagnostics, Chuncheon, Korea and AniGen, Bionnote, Hwasung, Korea) for NSP antibody detection.

Information of samples used for the FMD diagnosis and GWAS analysis
Identification of foot-and-mouth disease virus infected or resistant animals
We assumed that the animals from the infected farm were equally exposed to the risk of being infected by the circulating virus prior to vaccination. Animals that had antibodies for both SP and NSP were considered to be infected animals and those that produced antibody for only SP were considered to be the resistant animals.
Genome-wide association study and statistical analysis
Whole blood samples were collected to extract genomic DNA and the quality of DNA was adjusted for Chip assay. Illumina Bovine HD 770K SNP BeadChip including 777962 SNPs across the bovine autosomes and the X chromosome was used to genotype the animals. For the quality control, we excluded all SNPs with a miss rate of >5%, a minor allele frequency of <0.01 and a Hardy-Weinberg equilibrium test p-value of <10−6 using PLINK (Purcell et al., 2007). The SNP annotation information was used based on UMD 3.1 assembly version. In order to identify significantly associated SNPs with the trait, PERMORY (Pahl and Schäfer, 2010) was used for permutation test of GWAS case-control study with 1,000,000 random permutations. Permutation test is widely used for multiple test correction, since there are potentially high numbers of false positive findings in GWAS.
RESULTS AND DISCUSSION
We performed a GWAS of 96 female Holstein for FMDV resistance using the high density Bovine HD770K BeadChip. Out of 777,962 SNPs, a total of 624,532 were used for the association analysis after quality control. After PERMORY test, 11 SNP markers on 3 chromosomal locations showed the significant association with the FMDV resistance at p.adjust<0.05 (Figure 1). Three SNPs were located on chromosome 17, 7 on chromosome 22 and 1 on chromosome 15 (Table 2). Three SNPs on chr17 showed the most significant association and were located in the intergenic regions between Myosin XVIIIB (MYO18B) and seizure related 6 homolog (mouse)-like (SEZ6L). The MYO18B is known to be a tumor-suppressor gene and mutations in this gene are associated with lung cancer (Nishioka et al., 2002; Yokota et al., 2003; Tani et al., 2004; Yanaihara et al., 2004; Yokota and Kohno, 2004; Nakano et al., 2005). The Met430Ile polymorphic variant of SEZ6L has been previously linked with an increased risk of lung cancer in a human case-control population and the genetic and/or epigenetic alterations in SEZ6L might be involved in the development and/or progression in a subset of lung cancer (Nishioka et al., 2000; Gorlov et al., 2007). Among 7 significantly associated SNPs on chr22, 5 were spanning in the regions of CCDC36 (4 in introns and 1 in an exon) and 2 were in intergenic regions between CCDC36 and C22H3orf62. The SNP (rs110474439) in the exon of CCDC36 showed synonymous substitution. Due to the lack of knowledge on those genes, there are difficulties of understanding their function. However, ubiquitin specific peptidase 4 (USP4, proto-oncogene) that is 1 kb away from the C22H3orf62 plays an essential role in the negative regulation of the toll-like receptor/interleukin 1 receptor signaling-mediated innate immune response (Zhou et al., 2012). Although no SNPs in USP4 were significant at p.adjust<0.05 after PERMORY test, one (rs110159438) of them showed the association at p.adjust = 0.147 which was placed within top 20 markers. The SNP on Chr15 were located in an intron of DEAD box polypeptide 10 (DDX10), which is associated with leukemia (Nishiyama et al., 1999; Nakao et al., 2000; Morerio et al., 2006). Based on the known function of genes nearby the significant SNPs, we made inference that they might be involved in the functions related to innate immune responses. In contrast to adaptive immune system, induction of innate immune response by FMDV infection is still in a relatively early stage of understanding. One of major functions of the innate immune system is to act as a physical and chemical barrier to infectious agents (Janeway et al., 2001). The FMDV infects and replicates efficiently in epithelial cells (Summerfield et al., 2009). The epithelial surfaces of tissues that are in contact with the external environment, such as skin and the inner mucosa lining of the nose and lungs form a physical barrier which is very impermeable to most infectious agents and act as the first line of defense against invading organisms (Male, 2006). With respect to the respiratory airways and lungs, cilia movement helps to remove infectious agents and mucus traps infectious agents. The FMD resistant animals in this study seem to have ability to improve the innate immune responses to FMDV infection, even though further studies on the link between the genes and innate immune system are needed.
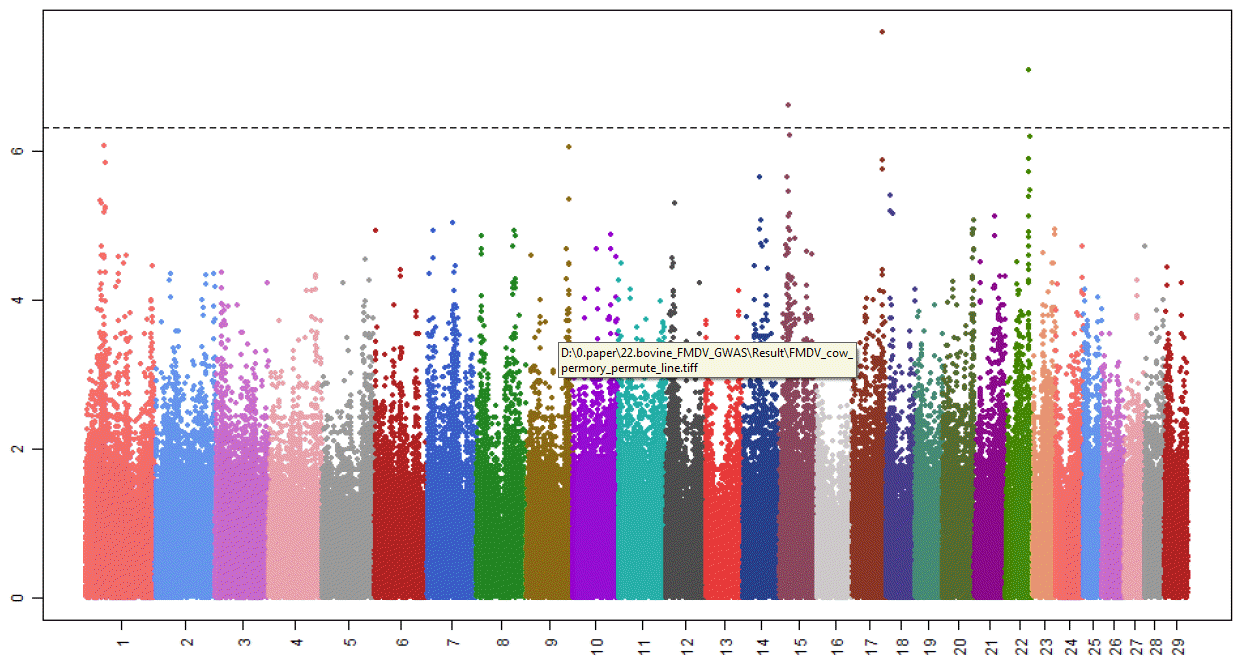
Manhattan plots showing the significant single nucleotide polymorphism (SNP) associated with the foot-and-mouth disease resistance. X-axis represents chromosome and Y-axis indicated −log10(p.adjust -value). Dots over the dotted line indicate significantly associated SNP.
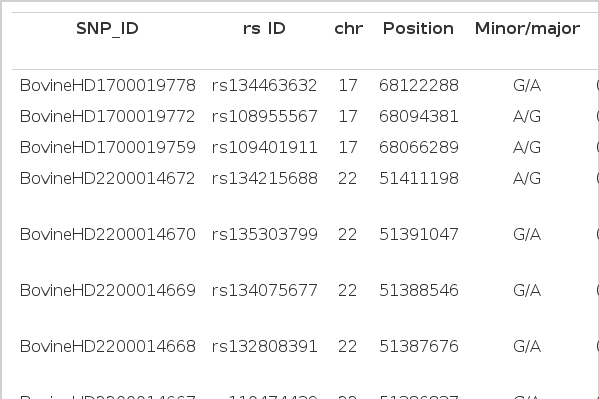
Eleven significant SNP markers associated with FMD resistant animals
Although we performed a GWAS study associated with the FMD resistance using high-density bovine SNP chip, we were aware of the limitation of this study such as small sample size and phenotyping without obvious clinical signs. Due to the eradiation policy, it was difficult to collect from large numbers of infected animals with clinical signs. After national vaccination program following the last outbreak, it was more difficult to collect infected samples. In order to collect even this small population, we had to make the assumption that the animals in the infected farm are equally exposed to the risk of being infected by the circulating virus during vaccination. Some animals co-housed with the infected animals had a chance of being infected after or before being vaccinated, even though they neither showed clinical symptoms nor the clear evidence of virus replication due to the protective immunity elicited from the vaccination. So the result of this study will require confirmation by further studies.
ACKNOWLEDGMENTS
This work was supported by grants from the Next-Generation BioGreen 21 Program (No. PJ008196), and from Cooperative Research Program for Agriculture Science & Technology Development (No. PJ006471), Rural Development Administration, Republic of Korea. We also thank Gyeonggi and Gangwon provincial veterinary services for collecting field samples.