A Whole Genome Association Study on Meat Quality Traits Using High Density SNP Chips in a Cross between Korean Native Pig and Landrace
Article information
Abstract
A whole genome association (WGA) study was performed to detect significant polymorphisms for meat quality traits in an F2 cross population (N = 478) that were generated with Korean native pig sires and Landrace dams in National Livestock Research Institute, Songwhan, Korea. The animals were genotyped using Illumina porcine 60k SNP beadchips, in which a set of 46,865 SNPs were available for the WGA analyses on ten carcass quality traits; live weight, crude protein, crude lipids, crude ash, water holding capacity, drip loss, shear force, CIE L, CIE a and CIE b. Phenotypes were regressed on additive and dominance effects for each SNP using a simple linear regression model, after adjusting for sex, sire and slaughter stage as fixed effects. With the significant SNPs for each trait (p<0.001), a stepwise regression procedure was applied to determine the best set of SNPs with the additive and/or dominance effects. A total of 106 SNPs, or quantitative trait loci (QTL) were detected, and about 32 to 66% of the total phenotypic variation was explained by the significant SNPs for each trait. The QTL were identified in most porcine chromosomes (SSCs), in which majority of the QTL were detected in SSCs 1, 2, 12, 13, 14 and 16. Several QTL clusters were identified on SSCs 12, 16 and 17, and a cluster of QTL influencing crude protein, crude lipid, drip loss, shear force, CIE a and CIE b were located between 20 and 29 Mb of SSC12. A pleiotropic QTL for drip loss, CIE L and CIE b was also detected on SSC16. These QTL need to be validated in commercial pig populations for genetic improvement in meat quality via marker-assisted selection.
INTRODUCTION
Consumers’ preference on pork has enabled to produce high meat in pig industry. To meet this ends, studies on genetics to identify genes or chromosomal regions (QTL) that are associated with meat quality have been performed, and lots of QTLs (6,347) for economically important traits have been reported in pig (http://www.animalgenome.org/QTLdb/).
Experimental crosses of two genetically divergent breeds, such as wild boar vs. European domestic breeds, or Meishan vs. western breeds were extensively used in QTL studies (Andersson et al., 1994; Rothschild et al., 1995, 2007; Janss et al., 1997; Bidanel and Rothschild, 2002; Hu et al., 2007). After the first report on genome-wide scan and detection of QTL for growth, length of small intestine, and fat deposition in pig (Andersson et al., 1994), many QTL studies using F2 cross populations were performed on growth and fat deposition (Knott et al., 1998; Paszek et al., 1999; Rohrer, 2000; Wada et al., 2000; Bidanel et al., 2001; Malek et al., 2001a; Kim et al., 2005), carcass traits (Andersson-Eklund et al., 1998; Rohrer et al., 1998), and meat quality (de Koning et al., 1999, 2001; Malek et al., 2001b). Recently, several QTL studies on purebreds for meat quality were also reported (Uemoto et al., 2008; Soma et al., 2011).
Korean native pig (KNP) were reported with good meat qualities such as better redness, less cooking loss and shear force than Yorkshire or Landrace breeds (Cho, 2006), and with good amounts of amino acid compositions that were possibly related to flavor and palatability (Hwang et al., 2004). Kim et al. (2007) and Choi et al. (2011) conducted genome-wide scans using microsatellite markers in an F2 cross population between KNP and Landrace to detect Mendelian and parent-of-origin QTL for growth, body composition, and meat quality traits.
Recently, with the developments of high throughput genotyping technologies such as next generation sequencing and high density porcine SNP chips, it is possible to genotype large amount of SNPs, e.g. with the porcine 64k Illumina Infinium assay, which enabled genome-wide association (GWA) study to detect QTL with high mapping resolution up to 1or 2 Mb confidence region (Fan et al., 2009; Gorbach et al., 2009; Onteru et al., 2009).
In this study, we performed whole genome scans to find significant sets of SNPs that were related to meat quality using the Illumina porcine 64k SNP chips in the KNP× Landrace F2 cross population that was used in Kim et al. (2007) and Choi et al. (2011).
MATERIALS AND METHODS
Animals and phenotypes
Data were collected from the QTL experimental population that were produced by crossing Korean Native (KNP) boars and Landrace (LN) sows at National Livestock Research Institute (NLRI), Songhwan, Korea. Ten F1 boars were randomly chosen for inter se mating up to six F1 sows to produce 38 full-sib F2 families resulting in 490 F2 progenies from the first (N = 281), second (N = 130), and third (N = 79) parities. The raising and management practices of the animals were described in Choy et al. (2002a; 2002b). The F2 individuals were slaughtered at the age of 201 to 251 d with the average of 214.8 d at the NLRI slaughterhouse. The average final (live) weight before slaughter was 91.6 kg with a range of 45.2 to 145 kg. Data measurements on ten carcass quality traits (live weight, crude proteins, crude lipids, crude ash, water holding capacity (WHC), drip loss, shear force, CIE L, CIE a, and CIE b, were described in detail in Choi et al. (2011).
Molecular data
Using the Infinium HD Assay Ultra Protocol (Illumina), all of the 490 F2 animals were genotyped with the porcine 64k SNP beadchips (Illumina Inc., USA). Raw data were visualized and analyzed with the Genome Studio software (Illumina). For subsequent data analysis, a subset of SNP was selected by removing the SNPs in sex chromosomes. Every SNP from the chip data was screened for the availability of GWA tests. Those SNPs on autosomal chromosomes were removed before WGA testing, which met the following three criteria; i) the number of genotype group with one or none (e.g. only AA genotypes and no AB or BB), ii) with a minor allele frequency less than 0.05, and iii) with the proportion of genotyped individuals less than 90%.
Statistical analysis
A SAS GLM procedure (SAS version 9.1) was used to preadjust the animal phenotypes before WGA testing. Sex, sire and slaughter stage were fitted as fixed effects for all meat quality traits, except the live weight, for which the effect of slaughter stage was left out of the model. Then, the residuals of each phenotype were regressed on the additive and dominance SNP effect for each SNP under a simple linear regression. In the model, SNP genotypes with AA, AB and BB were assigned as 1, 0, and -1 for the additive effect, and 0, 1, 0 for dominance effect, respectively. For significance threshold, 0.1% point-wise p value from F distributions was applied for each SNP test.
Among the significant SNPs, the best set of SNP markers for each trait were selected using the stepwise regression procedures (Neter et al., 1990), because some of the significant SNPs would yield redundant information due to linkage disequilibrium (LD) between closely linked SNPs, i.e. a non-random association between alleles of different SNPs. Inclusion and exclusion of each SNP out of the model was determined at p<0.001 level.
With the additive and/or dominance effects of the significant SNPs that were determined by the stepwise regression procedures, variation explained by each (jth) SNP
RESULTS AND DISCUSSION
Summary statistics for the ten meat quality traits were displayed in Table 1. The coefficient of variation (CV) for crude lipid was the greatest (96%), followed by for drip loss (87%). Among the color scores, CIE b had the greatest CV (43%), while the lowest CV was observed for crude protein (5%).

Summary statistics for 490 F2 phenotypes on meat quality in a KNP×LN cross population
A set of 46,865 SNPs was chosen from the 62,163 SNPs in the Illumina porcine beadchip (Table 2). The number of SNPs (6,622) was the greatest for Sus scrofa chromosome (SSC) 1, while SSC12 had the smallest number of SNPs (1,098). The SSCs 4, 7, 13, and 14 had more than 3,000 SNPs. The physical map with all of the available SNPs spanned about 2,424 Mb with an average distance of 44.6±133.5 Kb between adjacent SNPs. However, the average distances were different between chromosomes, ranging between 35.6 Kb in SSC17 and 87.3 Kb in SSC6. Only 75% of the SNPs that were embedded in the porcine chip were available in this study, partly due to ascertainment bias, i.e. the Illumina porcine 64k SNP beadchip was based on the genetics of other commercial breeds, not of Korean native pigs.
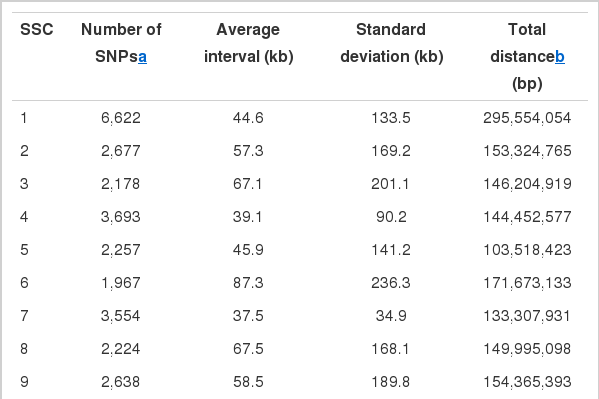
The numbers of available SNPs in the Korean native pig and Landrace F2 population and average interval distances between adjacent SNPs in all 18 Sus scrofa autosomes (SSC)
A total of 106 SNPs were detected in various chromosomes by the stepwise regression procedures, and the sets of SNPs for the ten carcass quality traits explained significant proportion of phenotypic variance, ranging between 31.8% for WHC and 65.6% for drip loss (Table 3). For live weight, twelve SNPs (QTL) were found on SSCs 1, 2, 5, 7, 13, 14, 15, and 16, of which multiple QTL were located on SSCs 5, 7, 13, and 14 (Table 3). Murani et al. (2005) performed association analyses in Pietrain and Duroc×Pietrain populations, and found QTL for ham weight on SSC2 (EPOR gene at 52 Mb), which was closely located to the QTL (47 Mb) in this study. Soma et al. (2011) performed genome-wide scans in Duroc and detected QTL for carcass length and body length at 102 cM and 105 cM of SSC13 that were in close distance to the QTL (96 Mb) for live weight in this study (Table 3).
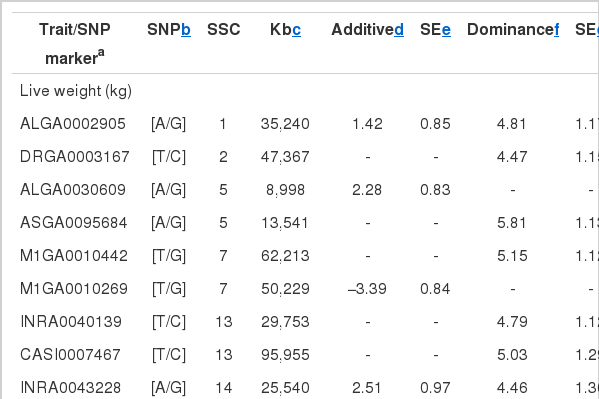
Identities, positions of the SNPs with significant effects on meat quality traits in a Korean native pig and Landrace cross population
Eleven significant QTL for crude proteins were detected, among which multiple QTL, i.e. two, four and two QTL were detected on SSCs 1, 12, and 16, respectively (Table 3, Figure 1). Especially the three of the four QTL on SSC12 were clustered between 20 and 29 Mb. In our previous linkage mapping results using the same population as in this study, a QTL for crude protein was detected at 0 cM of SSC1 (Choi et al., 2011). In the proximal region (6 Mb), we detected one QTL for the same trait (Table 3).
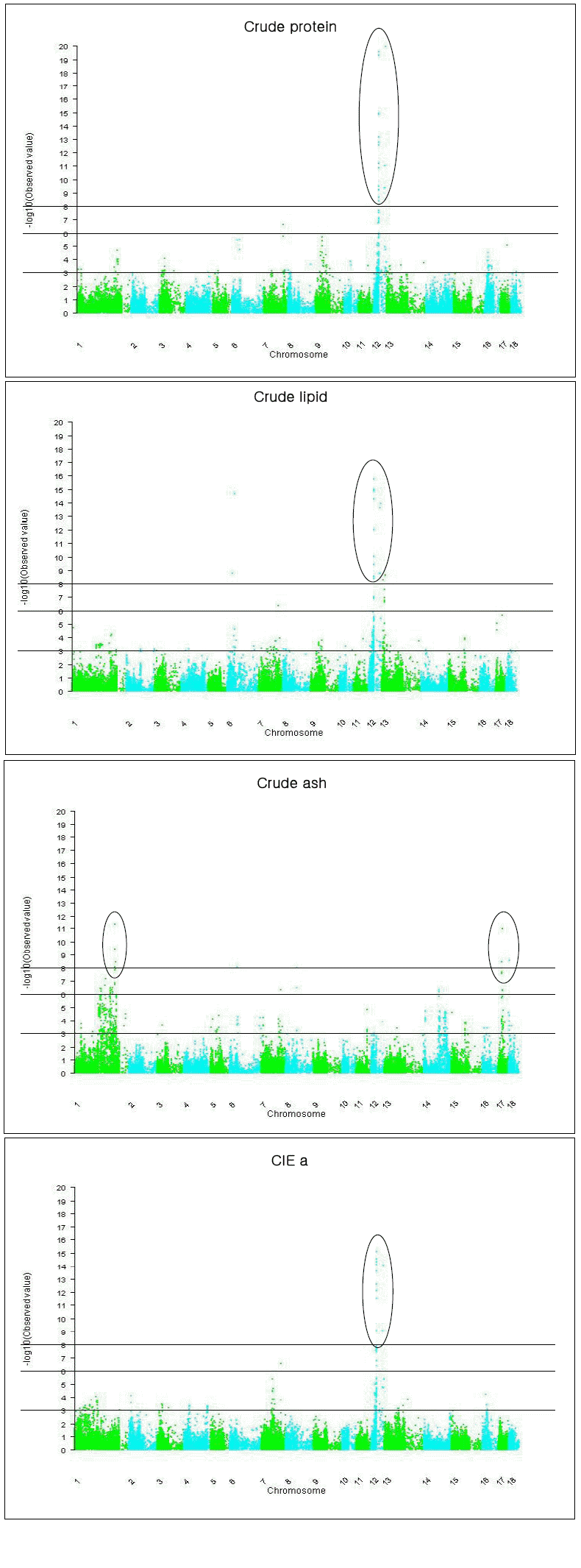
Test statistic profiles for crude protein, crude lipid, crude ash and Cie a under the additive and dominance model for each SNP. Circles indicate highly significant QTL. Y axis indicates −log10(p-value under the null hypothesis of no SNP effect).
Ten QTL for crude lipid were detected on SSCs 6, 7, 12, and 13, of which eight QTL were located on SSC6 and SSC12. The detected QTL explained significant proportion (57%) of phenotypic variation (Figure 1, Table 3). Stearns et al. (2005) detected a QTL for fat % at 19 cM of SSC6 in a cross population of Berkshire and Duroc. We identified one QTL for crude lipid at 31 Mb of the same chromosome (Table 3). Sanchez et al. (2007) detected a QTL for fatty acids (C16:1, Palmitoleic) at 40 cM of SSC12 in a Duroc×Large White cross, which was located distal to the crude lipid QTL at 29 Mb in this study. Munoz et al. (2007) detected five QTL for fatty acid composition in between 1 and 34 cM, and three QTL at between 68 and 76 cM of SSC12 in an Iberian and Landrace F2 cross, in which five QTL for crude lipid were detected in this study (Table 3). Clop et al. (2003) also reported two QTL for unsaturated index and linolenic acid at 34 cM of SSC6 and 31 cM of SSC 12, respectively, in an Iberian×Landrace F2 cross population. In the similar regions, multiple QTL for crude lipid were identified in this study (Table 3).
For crude ash, 12 QTLs were detected on eight SSCs, among which the four QTL on SSC17 were clustered at 21 to 26 Mb regions (Table 3). Ovilo et al. (2002) reported QTL for Haematin in an Iberian×Landrace F2 cross population at 109 cM of SSC4 and 87 cM of SSC7, respectively. Haematin is an iron-containing compound which might be related to the crude ash. We detected two QTL for crude ash at 109 Mb and 101 Mb of the respective chromosomes (Table 3).
Ten QTL for WHC were identified on six SSCs, among which multiple QTL were observed on SSCs 2, 9, and 13 (Table 3). Malek et al. (2001b) detected two QTL for the trait on SSC2 and SSC13 in a cross population of Berkshire and Yorkshire. The QTL locations were at 71 cM and 33 cM, respectively, that were close to the QTL in this study (Table 3). Murani et al. (2005) detected a drip loss QTL in EPOR gene region (on SSC2, 52 Mb) in Pietrain, and Duroc×Pietrain cross population. Usually drip loss and WHC are considered to be highly correlated each other, and we detected one QTL for WHC at 61 Mb of the same chromosome.
Nine QTL for drip loss were found on SSCs 4, 5, 9, 10, 12, 14, 15 and 16, respectively (Table 3). Jennen et al. (2007) identified a candidate gene for drip loss, ATF4, at 50 Mb of SSC5, which was close (45 Mb) to the QTL in this study. Thomsen et al. (2004) found QTL for drip loss on SSC5 and SSC9 in a Berkshire and Yorkshire cross population. In the same chromosomes, two QTL for the trait were detected (Table 3). Li et al. (2011) reported that a SNP in DLX3 gene at 24 Mb of SSC12 was associated with drip loss in the same population as in this study, while a QTL for drip loss was detected at 28 Mb of SSC12 (Table 3). In a Duroc purebred population, Li et al. (2010) found a QTL for the trait on SSC15 (63 cM), and Murani et al. (2005) also reported that the TTN gene (at 79 Mb, Ensembl database) on SSC15 was associated with drip loss in a Duroc×Pietrain cross population. In this study, a QTL for drip loss was detected at 71 Mb (Table 3).
Eleven QTLs for shear force were detected on eight SSCs, of which multiple QTL were located on SSCs 1, 3 and 13 (Table 3). Choi et al. (2011) reported a QTL for shear force at 19 cM of SSC15 that was detected under a line-cross model based linkage analyses using the same population as in this study. Thomsen et al. (2004) also detected one QTL for tenderness at 13 cM of the same SSC in a Berkshire×Yorkshire cross population. In this study, one QTL for shear force was detected at 9 Mb of the same chromosome (Table 3). Murani et al. (2005) reported the PDGFRA gene on SSC8 (35 Mb) in a Duroc cross population, and the NME1 gene on SSC12 (25 Mb) in Duroc×Pietrain crosses, which were associated with shear force. Results from Li et al. (2011) and Choi et al. (2011), in which the same KNP×LN cross population was used as in this study, included two QTL for shear force on SSC12 and SSC15, respectively, which were closely located to the two QTL (23 Mb, 9 Mb) on the respective SSCs in this study (Table 3).
Most QTL for meat color traits (CIE L, a, b) were found in many chromosomes, especially on SSCs 1, 2, 4, 5, 11, 12, 14, and 16 (Table 3). In some SSC regions, many QTL for meat color were reported; on SSC4 and SSC7 for Minolta a (Ovilo et al., 2002), on SSC2 and SSC14 for Hunter L, and on SSC5 for Hunter b (Roher et al., 2005), on SSC2 for A value (Streans et al., 2005), on SSC4 for color 24hSM (Ma et al., 2009), on SSC1 for L value, on SSC14 for A value, and on SSC4 and SSC5 for B value (Li et al., 2010), and on SSC14 for CIE L (Li et al., 2011). Our study also detected QTL for CIE a, b, and L in these QTL regions (Table 3). Duthie et al. (2008) reported an epistatic effect of OPTO-STAR for CIE L and b that was located on SSC4, which also resided in the similar region to the QTL for CIE L and b in this study (Table 3). The QTL for CIE a that was located at 27 Mb of SSC12 had the greatest statistical evidence of this QTL study (Table 3, Figure 1).
On SSC12, many QTL were clustered between 20 and 29 Mb for crude protein, crude lipid, drip loss, shear force, CIE a and CIE b (Table 3), suggesting that more than one gene reside in the regions. Also, on SSC16 and SSC17, SNPs with pleiotrophic effects were detected, e.g. ALGA0090076 on SSC16 for drip loss, CIE L, and CIE b (Table 3).
Previously, we performed genome-scans to detect QTL for meat quality using the same KNP×LN cross population as in this study (Choi et al., 2011; Li et al., 2011). However, very limited QTL were confirmed in this study, partly because different QTL mapping approaches were applied with the use of different set of markers. In this study, WGA was applied using high density SNP chips, while Choi et al. (2011) performed QTL analysis with limited numbers of microsatellite markers under the interval mapping models that were based on breed-cross designs with Mendelian and non-Mendelian inheritance mode of the QTL. Also, Li et al. (2011) performed association analyses using limited number of SNPs of candidate genes.
CONCLUSION
Selection for high growth and lean meat has been extensively implemented in pig industry. However, recent genetic improvement has been focused on better meat qualities. In this study, lots of QTL (108 SNPs) were detected for meat quality, providing a set of genetic markers to implement marker-assisted selection (MAS). Several QTL clusters were identified on SSCs 12, 16, and 17, as well as QTL with pleiotrophic effects, e.g. the QTL for drip loss, CIE L and CIE b on SSC16. Characterization of these QTLs is needed to find causal mutations around the QTL, which would enable MAS to be implemented with greater efficiency and accuracy.
ACKNOWLEDGEMENT
This work was supported by grants from 2-5-13 Agenda research (PJ006707) from the National Institute of Animal Science and a grant (PJ008068, PJ008089) from the Next Generation BioGreen 21 Program, Rural Development Administration, Republic of Korea and the support of “Cooperative Research Program for Agriculture Science & Technology Development (PJ009103)”, the Rural Development Administration, Republic of Korea.