



INTRODUCTION
Cattle genetic resources are among the most valuable assets both in developed and developing nations. Human societies have, for at least 10,000 yr, recognized the importance of these assets and have been engaged in the domestication to meet a variety of needs (Loftus et al., 1994, Bradley et al., 1996). Cattle genetic resources make a major contribution to human requirements for food in the form of meat, milk and milk products as well as traction power. Worldwide, several cattle breeds have contributed to these agricultural products since domestication. Although cattle breeds provide these valuable products, a number of breeds have become extinct over the past half century. For instance, there is a general consensus that among the 7,600 livestock breeds registered in the FAOŌĆÖs global database, 190 have become extinct in the past two decades and a further 1,500 are considered at risk of extinction (FAO, 2009).
The loss of genetic variation caused by population reduction is another important concern in cattle genetic resources management. Genetic diversity has been defined as the variety of alleles and genotypes present in a population and that is reflected in morphological, physiological and behavioral differences between individuals and populations (Frankham et al., 2002). It has long been known from studies with domesticated animals that the loss of genetic variation has a variety of harmful effects on survival and growth rate (Lande, 1988; Falconer, 1996). A loss of genetic variation can also limit evolutionary potential (Frankham et al., 1999), and reduce the ability of a population to cope with newly introduced pathogens and parasites (OŌĆÖBrien and Evermann, 1989).
The Hanwoo cattle are the most important cattle in the Korean peninsula, four strains of Hanwoo namely Korean Black, Jeju Black, Korean Brindle and Korean Brown are recognized from the traditional population, showing phenotypic and genetic differences (Choi, 2009). Hanwoo is the indigenous cattle of Korea, and the history of this breed dates back at least 5,000 yr. With regard to Hanwoo breed origin, several hypotheses are available (Han, 1996; Lee and Pollack, 2002; Yoon et al., 2005). The common hypothesis is that Hanwoo cattle originated from a cross between Bos indicus and Bos taurus subspecies (Han, 1996). The population size of KB, JEB and BRI populations have been considerably reduced over the past decades, mainly due to the exclusion of these populations from breed improvement programs. We assume that reduction in population size encountered by these populations could have had effect on their genetic diversity and population demographic history. Understanding the patterns of genetic diversity and demographic history of these cattle would help in implementing more informed breed management strategies. Here, we investigated the patterns of mitochondrial control region sequence variation in these cattle populations in order to describe the population genetic diversity, genetic structure between populations and changes in population size. First, we evaluated to determine whether or not these cattle encountered a reduction in population size. Second, population reduction detected by mismatch distribution analysis and neutrality tests was further evaluated if this reduction could have had effect on the genetic diversity of each population. The objective of this study was to examine the extent to which reduction in Korean Black, Jeju Black and Korean Brindle cattle populations influenced genetic diversity, population structure and demographic history using complete mitochondrial DNA control region sequences.
MATERIALS AND METHODS
In total, 56 samples were collected from three Korean native cattle populations viz, Jeju Black (JEB) (n = 19), Korean Black (KB) (n = 18) and Korean Brindle (BRI) (n = 19). DNA samples of Jeju Black cattle were otainted from Subtropical Animal Experimental Station (National Institute of Animal Science) in Jeju island, and DNA samples of Korean Black and Brindle were obtained from Institute of Livestock and Veterinary Research (Chungbuk province). We included 27 mtDNA sequences of Korean Brown (BRO) accession numbers AF499238-AF499264 (Lee et al., 2002) from the GenBank database.
Mitochondrial DNA amplification and sequencing
The complete control region of mtDNA (910 bp) was amplified using forward primer L15737: 5ŌĆ▓-CTGCAGTCTCACCATCAACC-3ŌĆ▓ and reverse primer H992: 5ŌĆ▓-GATTATAGAACAGGCTCCTC-3ŌĆ▓ (Anderson et al., 1982). The reactions were carried out using 0.2 ╬╝g of mtDNA, 0.4 ╬╝M of each primer, 0.25 mM dNTP, 10├Ś reaction buffer contains Tris-HCl (pH 9.0), PCR enhancers, (NH4)2SO4, 20 mM MgCl2 and 1 U Taq DNA polymerase in a 25 ╬╝l reaction volume. Amplifications were carried out as follows: 3 min at 94┬░C for initial denaturation followed by 35 cycles each of 1 min at 94┬░C, 30 s at 58┬░C, 90 s at 72┬░C and a final extension step of 5 min at 72┬░C.
The PCR products were purified using SolGentŌäó PCR Purification Kit, following the manufacturerŌĆÖs instructions. The PCR products were then sequenced using both PCR primers and an internal primer L16161:5ŌĆ▓-ATTACCATGCCGCGTGA-3ŌĆ▓. Sequencing was performed using a BigDye Cycle Sequencing Kit on an ABI-3730XL DNA Analyzer. Sequence variations in the control region of mtDNA from Korean cattle populations were defined by comparison with a standard bovine sequence accession no.V00654 (Anderson et al., 1982).
Statistical analyses
All sequences were checked for ambiguous bases and edited manually using BioEdit V7.0 (Hall, 1999). The complete mtDNA control region sequences were aligned using CLUSTALW (Thompson et al., 1994) as implemented in MEGA (Tamura et al., 2011). All positions containing gaps and missing data were excluded from the analysis.
Mismatch distribution
To gain insight into whether a reduction in population size has occurred in the past, we examined the distribution of the observed number of differences between pairs of haplotypes, The distribution is usually multimodal in samples drawn from populations at demographic equilibrium, as it reflects the highly stochastic shape of gene trees, but is expected to be unimodal in populations that have undergone a recent demographic expansion (Rogers and Harpending, 1992). The method assumes a sudden stepwise expansion model from a population of N0 to N1 individuals, t generation ago followed by demographic equilibrium. Three parameters were estimated with coalescent simulations, Žä = 2╬╝t, ╬Ė0 = 2N0╬╝ and ╬Ė1 = 2N1╬╝, where ╬╝ is the mutation rate for the locus. The variables ╬Ė0 and ╬Ė1 are estimates of population sizes before and after an expansion, respectively, and Žä is the time since the expansion occurred. The validity of the estimated stepwise expansion model was tested. We used the sum of square deviations (SSD) between the observed and the expected mismatch as a test statistic; all calculations were done using arlequin3.11 (Excoffier et al., 2005).
Neutrality test
We performed FuŌĆÖs (1997) Fs test by comparing Fs statistics against a distribution generated from 1,000 random samples (Excoffier et al., 2005). This test is based on the infinite-allele model. The statistic Fs tend to be negative under an excess of recent mutations and a significant negative value will be taken as evidence of population growth using arlequin We also estimated TajimaŌĆÖs (1989) D statistics, and the significance of the D statistic was tested by generating random samples under the hypothesis of selective neutrality and population equilibrium, using a coalescent algorithm (Excoffier et al., 2005).
Level of genetic diversity and structure
Patterns of sequence variation, number of variable sites, mitochondrial haplotype diversity (h) and nucleotide diversity (ŽĆ) were estimated, all measures were calculated using the program DnaSP Ver 5.10.01 (Rozas et al., 2003). The extent of population genetic differentiation was quantified by FST statistic using arlequin (Excoffier et al., 2005). Genetic structure of Korean cattle populations was further assessed using the analysis of molecular variance (AMOVA) as implemented in arlequin. Significance tests were performed using 1,000 permutations. AMOVA estimate genetic structure indices using information on the allelic content of haplotypes as well as their frequencies (Excoffier et al., 1992).
RESULTS
Sequence variation
In the alignment of the sequences, 3, 15, 19 and 33 variable sites were detected defining 3, 9, 12 and 21 haplotypes in KB, JEB, BRI and BRO, respectively. All the variable sites represent substitution mutation. A reduced number of variable sites was detected in KB cattle. The BRI, JEB and KB depicted 3, 1 and 3 singleton and 16, 14 and 2 parsimony informative variable sites, respectively. In BRO cattle, 20 singleton and 13 parsimony informative variable sites were detected.
Mismatch distribution and neutrality tests
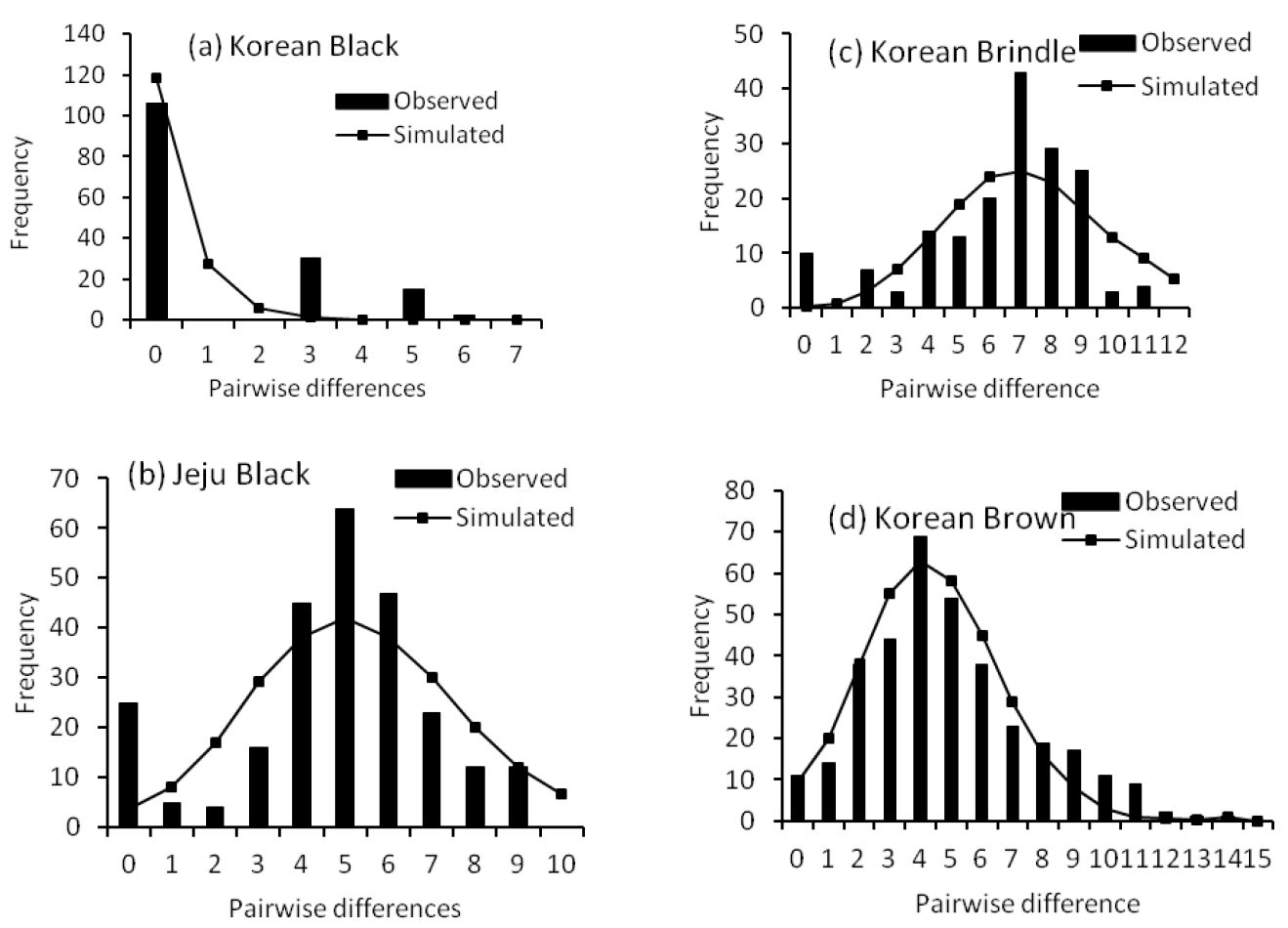
As depicted in Figure 1a, the mismatch distribution did not reflect a signature of demographic population expansion for KB, the analysis revealed a significant SSD (p<0.05) value that rejected the sudden expansion model (Table 1), suggesting either a population in demographic equilibrium or a population decline. There were patterns of demographic population expansion for JEB and BRI cattle (Figure 1b and c). However, the p-values of the SSD test was significant, the observed mismatch distributions of BRI and JEB did not match the expected distributions under the sudden expansion model (Table 1). On the other hand, mismatch distribution analysis revealed that the BRO cattle population showed the genetic signal of a sudden population expansion, exhibiting unimodal mismatch distribution (Figure 1d) and the p-values of SSD was not significant (Table 1).
The results of FuŌĆÖs and TajimaŌĆÖs neutrality tests for the Korean cattle populations are presented in Table 1. The two tests of neutrality were not statistically significant for KB, BRI and JEB cattle populations. The results suggested that these populations are in genetic equilibrium or in decline. The tests did not support the patterns of a population expansion event that were generated by mismatch distribution analysis for KB and BRI. The TajimaŌĆÖs and FuŌĆÖs neutrality tests were both significant for BRO population, in support of the mismatch distribution analysis.
Level of genetic diversity and structure
The estimated nucleotide diversity in the total population was 0.004. The estimated within population nucleotide diversity varied from 0.001 to 0.005 (Table 2). Haplotype diversity in the total population was 0.87. Within population, KB showed the lowest haplotype diversity of 0.368 while the highest was 0.971 in BRO (Table 2).
The FST values computed for pairwise comparisons revealed low to moderate genetic differentiation (Table 2). There was no genetic differentiation between BRO and BRI (FST = 0.014) and BRI and JEB (FST = 0.005) populations. This analysis revealed low but highly significant differentiation between BRO and JEB. As depicted in Table 2, moderate significant differences between BRO and KB, JEB and KB, KB and BRI were detected. Relatively, the KB population was genetically distant from the rest of the cattle populations studied. The level of population genetic structure assessed by AMOVA indicated that the proportion of genetic variation among the Korean cattle populations was 7.6% (p<0.001), and about 92.4% of the variance in the samples was attributable to within population variation.
DISCUSSION
This paper describes the first attempt to evaluate the influence of the recent population reduction on genetic diversity, population structure and demographic history of three Korean cattle populations. Understanding the patterns of genetic diversity and structure would allow not only a better understanding of the demographic and evolutionary history of the cattle populations, but also such information will help in implementing more informed breeds management strategies.
Mismatch distribution and neutrality tests
Populations at demographic equilibrium or in decline should present a multimodal distribution of pairwise differences, while populations that have experienced a sudden demographic expansion should display a unimodal distribution (Slatkin and Hudson 1991; Rogers and Harpending 1992). An excess of singleton variable sites may be an evidence for population expansion (William et al., 1995). Excess of singleton variable sites were detected in BRO, suggesting population expansion. In fact, the expansion scenario of BRO population revealed by mismatch distribution was also supported by FuŌĆÖs Fs and TajimaŌĆÖs D tests. Unlike BRO, a few singleton variable sites were detected in KB, JEB and BRI cattle populations. The goodness-of-fit of the observed mismatch distribution to the simulated model of population expansion tested with SSD was significant for KB, JEB and BRI cattle populations, the test statistic rejected the population expansion model. Furthermore, FuŌĆÖs Fs statistic, which was devised specifically to detect population expansion and is more sensitive to the presence of singletons, was not significant for these cattle populations. TajimaŌĆÖs neutrality test also did not support population demographic expansion for these populations. In general, the neutrality tests were suggestive of population decline for KB, JEB and BRI.
Level of genetic diversity and structure
Based on mtDNA sequence variation, the estimates of genetic diversity observed in the KB cattle were low in comparison to the others cattle populations studied. In KB, only 3 haplotypes and a low haplotype and nucleotide diversity were detected. The observed mtDNA diversity in KB is the characteristic of population that has undergone a reduction in population size. Such populations are expected to show a rapid increase in the proportion of identical or closely identical haplotypes (Hoelzel et al., 1993; Weber et al., 2000). This phenomena arises because some haplotypes are lost through genetic drift so that subsequent animals are descended from a few surviving female ancestors. The extreme reduction in mtDNA diversity in the KB is largely attributed to a reduction of its population size that followed the exclusion of this breed from registration as a pure breed for breed improvement. Generally, the focus of cattle genetic improvement has been on BRO for several years and there was little attention given to BRI, KB and JEB cattle populations in previous years.
It is typically considered that a small or bottlenecked population lacks genetic diversity (Amos and and Balmford, 2001; Spielman et al., 2004). The BRI and JEB cattle have been reduced in population size, however, they maintained genetic diversity which is comparable to BRO cattle whose population size has not declined. This study reveals that the mtDNA diversity of BRI and JEB did not reflect their population size. Within population genetic diversity is thought to reflect population size, ecology, history and ability to survive. In another empirical investigation, using a comprehensive collection of polymorphism data sets covering approximately 3,000 animal species, Bazin et al. (2006) reported that the widely used mtDNA marker does not reflect species population size or ecology; no correlation between mtDNA diversity and species abundance could be detected. Several factors can of course affect genetic diversity, including population structure (Cherry and Wakeley, 2003), population bottlenecks (OŌĆÖBrien, 1994), natural selection and mating systems (Hedrick, 2005). The influence of these factors can complicate an attempt to interpret the genetic diversity of any population in terms of population size (Amos and Harwood, 1998), as we have observed in BRI and JEB cattle populations.
Analysis of molecular variance showed a significant population differentiation (FST), estimates of between population variability was moderate for some populations, indicating that these populations may be different from each other. Additionally, the partitioning of genetic diversity with AMOVA suggested that the majority of the genetic variation can be attributed to within populations. Among populations variability was approximately 7.6% of the total variability.
Several factors, including human preferences have led to the extant KB, BRI and JEB facing a decline in population size. There is a real risk that if the trend towards a decline in numbers is not reversed, genetic consequences related to genetic drift and inbreeding will further impoverish most of the genetic diversity in these cattle populations. Appropriate conservation strategies should be set in place for the survival of these cattle. Conservation objectives may range from maintaining genetic diversity, avoiding extinction to the extent of preserving the breeds as heritage or cultural identity of the local communities keeping them.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







