Detection of QTL for Carcass Quality on Chromosome 6 by Exploiting Linkage and Linkage Disequilibrium in Hanwoo
Article information
Abstract
The purpose of this study was to improve mapping power and resolution for the QTL influencing carcass quality in Hanwoo, which was previously detected on the bovine chromosome (BTA) 6. A sample of 427 steers were chosen, which were the progeny from 45 Korean proven sires in the Hanwoo Improvement Center, Seosan, Korea. The samples were genotyped with the set of 2,535 SNPs on BTA6 that were imbedded in the Illumina bovine 50 k chip. A linkage disequilibrium variance component mapping (LDVCM) method, which exploited both linkage between sires and their steers and population-wide linkage disequilibrium, was applied to detect QTL for four carcass quality traits. Fifteen QTL were detected at 0.1% comparison-wise level, for which five, three, five, and two QTL were associated with carcass weight (CWT), backfat thickness (BFT), longissimus dorsi muscle area (LMA), and marbling score (Marb), respectively. The number of QTL was greater compared with our previous results, in which twelve QTL for carcass quality were detected on the BTA6 in the same population by applying other linkage disequilibrium mapping approaches. One QTL for LMA was detected on the distal region (110,285,672 to 110,633,096 bp) with the most significant evidence for linkage (p<10−5). Another QTL that was detected on the proximal region (33,596,515 to 33,897,434 bp) was pleiotrophic, i.e. influencing CWT, BFT, and LMA. Our results suggest that the LDVCM is a good alternative method for QTL fine-mapping in detection and characterization of QTL.
INTRODUCTION
Meat and carcass quality of Korean native cattle, Hanwoo, are of great value and thus of great concern to Korean consumers. Among carcass quality traits, carcass weight, backfat thickness, longissimus dorsi muscle area, and marbling score are the most influential on beef profit in Hanwoo industry.
Hanwoo breeding programs that were implemented using Animal Model has enabled significant genetic progress in carcass quality. For example, the estimates of annual gain over the last 10 yr were 8 kg for CWT and 2.9 cm2 for LMA (NIAS, 2009). However, genomic prediction at young stage can further accelerate genetic gain, because prediction errors at breeding age would be reduced by exploiting information on the value of every chromosomal fragment and on the transmission of chromosome fragments from parents to selection candidates (Garrick, 2011).
Meuwissen et al. (2002) proposed a fine-mapping method that was based on the evolutionary history of adjacent markers under a coalescent model. In the mapping approach, QTL variance is estimated using a matrix of covariances among marker haplotypes flanking the QTL. If the haplotypes are similar, e.g. identical alleles between the haplotypes, there is an increased chance that the QTL alleles in the haplotypes that were descended from unobserved founders of the population are identical by descent (IBD), which was called linkage disequilibrium (LD) mapping. If the common ancestors occur within the known pedigrees, then the IBD probability between individuals can be calculated by investigating inheritance patterns of the marker alleles from the parents, i.e. by linkage analysis (LA).
Kim and Georges (2002) reported a modified version of Meuwissen et al. (2002) with the addition of a hierarchical haplotype clustering step in the QTL fine-mapping analysis, which clustered haplotype groups that were based on kinship relatedness and effectively solved numerical computation problems, e.g. singularities in the haplotype covariance matrix between similar haplotypes (Druet et al., 2008).
Chromosomal regions, genes, and their specific alleles on BTA6 that were associated with carcass traits in Hanwoo have been reported (Lee et al., 2008; Lee et al., 2010a; b).
The aim of this study was to further refine the QTL region on BTA6, and to search for candidate genes affecting carcass qualities in Hanwoo by applying the LDVCM method of Kim and Georges (2002).
MATERIALS AND METHODS
Animals, phenotypes and molecular data
The steers (N = 427) with phenotypes and molecular data were chosen among the progeny of candidate bulls for progeny testing in the Hanwoo Improvement Center of National Agriculture Cooperative Federation in Seosan, Chungnam province, Korea. The data set comprised 45 sires and their 427 steers that were born between spring of 2005 and fall of 2007. The number of steers for each of the 45 paternal sire families ranged from six to 13 with the average of eight steers. Four carcass quality traits were chosen that were measured in the populations; carcass weight after slaughter (CWT), backfat thickness (BFT), longissimus dorsi muscle area (LMA), and marbling score (Marb). Details about raising and management of the experimental population, data recording, and summary statistics for the traits were described in Li et al. (2011).
A set of 2,535 SNPs comprising a dense marker map covering BTA6 that were embedded in the Illumina Bovine SNP50K BeadChip were chosen (Matukumalli et al., 2009). Details on the SNP genotyping experiments were described in Lee et al. (2010a). A total of 1,855 phased SNPs on the BTA6 were selected and genotyped for the 427 steers (Li et al., 2011). Average minor allele frequency and observed heterozygosity (±SE) across all the SNPs was estimated as 0.27±0.14 and 0.36±0.13, respectively.
QTL analysis
A QTL fine interval-mapping approach was applied, which exploited linkage and linkage disequilibrium between individuals in both the observed and unobserved pedigrees, respectively (Kim and Georges, 2002; Blott et al., 2003). Firstly, the linkage phases of all sires and sons were determined by the approach of Druet and Georges (2010). Then, IBD probabilities (ϕp) at the midpoint of each SNP interval (p) were computed for all pairs of haplotypes conditional on the identity-by-state status of flanking markers (Meuwissen and Goddard, 2001). A dendrogram was generated by using the unweighted pair group method with arithmetic mean (UPGMA) hierarchical clustering algorithm with 1−ϕp as the distance measure at QTL location (p). Starting at the ancestral node and sequentially descending into the dendrogram, all possible combinations of haplotype clusters were analyzed in place of individual haplotypes. This process identified the set of nodes at which the likelihood of the data were maximized.
To jointly exploit LD and LA information, the following mixed linear was used:
Where y is the vector of phenotype records of all sons, b is a vector of fixed effects, which in this study reduces to the overall mean. X is the incidence matrix relating fixed effects to individual sons. h is the vector of random QTL effects corresponding to the defined haplotype clusters. Zh is an incidence matrix relating maternal haplotypes of sons and sire haplotypes to individual sons. s is the vector of random individual polygenic effects (sire model). Zs is a diagonal incidence matrix relating individual polygenic effects to individual sires. e is the vector of individual error terms. For each trait, appropriate fixed factors or covariates were fitted in the models (p<0.05) using a GLM procedure in SAS (SAS 9.1, SAS Institute Inc., Cary, NC, USA). Two fixed effects was fitted in the models; year and season of birth (5 levels) and region where the steers were born (40 levels), and one covariate, slaughter age, was also fitted.
The models were tested on each midpoint of each SNP interval on the BTA6. Likelihood ratio tests (LRT) were applied by comparing the maximum likelihoods between the full model with the testing haplotype clusters and the reduced model without the QTL effect:
The LRT test statistics approximately followed chi-squared distributions with 1 degree of freedom (Visscher, 2006). For significance threshold, 0.1% comparison-wise p value was applied for QTL detection.
RESULTS AND DISCUSSION
A total of 47 significant signals (SNPs) were detected, for which 13, 11, 21, and two SNPs were associated with CWT, BFT, LMA, and Marb, respectively (Table 1, Figure 1). The significant signals were grouped into 15 QTLs, comprising five, three, five, and two QTL for the respective traits (Table 1).
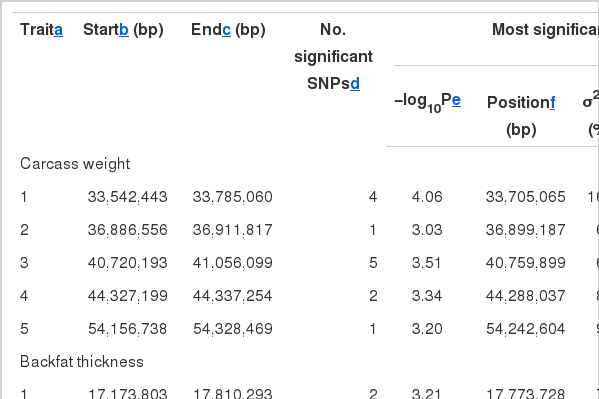
The QTL and SNP position that were detected for carcass quality traits on BTA6 in Hanwoo

Test statistic profiles along the BTA6 using the LDVCM analysis for carcass weight (CWT), backfat thickness (BFT), longissimus dorsi muscle area (LMA) and marbling score (Marb). The X-axis indicates position of chromosome 6, and the Y-axis indicates comparison-wise p values of the test-statistic of SNPs against the null hypothesis of no QTL.
In general, compared with Li et al. (2011), in which QTL analyses were performed under four different LD mapping models using the same phenotypes and genotypes as in this study, the LDVCM analyses gave similar results on QTL locations. However, the LDVCM outperformed in power in detecting QTL, i.e. only 12 QTL were detected in Li et al. (2011). This result may be partly due to genetic characteristics of Hanwoo breed. In Holstein-Friesian dairy cattle, high LDs, e.g. r2>0.62, were detectable within 10 kb distances between SNPs, suggesting that 300,000 evenly spaced SNPs are sufficient to cover the genome to exploit LD for QTL fine-mapping (Khatkar et al., 2008). In Hanwoo, however, LDs between linked SNPs were much weaker, compared to other western commercial breeds (Lee et al., 2010a). For this reason, the single marker regression analysis that was applied in Li at al. (2011) would cause less power in detecting QTL, because there is less chance that a QTL is closely located to one SNP marker with high LD. However, the LDVCM analysis, which was based on haplotypes and thus LD information was exploited from multiple SNPs flanking the QTL, would enable detection of QTL with greater power (Meuwissen and Goddard, 2001). Further, the LDVCM analysis exploited LD information within the observable pedigrees, i.e. inheritance of marker alleles from sires to progeny, as well as LDs at a population-wide level between chromosomes of sires and maternal chromosomes of steers (Blott et al., 2003). Thus, the LDVCM method would be suitable in QTL mapping efficiency when marker densities was limited and a structured population was used, e.g. paternal halfsib pedigrees.
Fifteen chromosomal regions (QTL) were identified (Table 1) and many significant signals (SNPs) were observed in the QTL regions (Figure 1). The signals may be due to high LDs with the causal mutation for the trait. Another reason of the clustering of significant SNPs might be due to the chromosomal regions harboring groups of co-regulated genes that are related to restriction sites or transcription factor binding sites (Sun et al., 2006). Therefore, even if more than one significant signals (SNPs) were detected within ±1 Mb region (2-Mb QTL interval), only one QTL was declared. The QTL can be accessed in the Ensembl (Hubbard et al., 2009).
Half of the significant signals (11 out of 21) that were associated with LMA were located on the distal region (110,285,672 to 110,633,096 bp) (Table 1, Figure 1). The QTL region was close to one gene, CLNK, which encodes cytokine-dependent hematopoietic cell linker. In particular, a signal (BTB-00280923 ~ Hapmap44204-BTA-117480) that was located at 110,360,573 bp was the most significant among all the tests of the four traits (p<10−5, Table 1).
In the proximal region flanking 33,596,515 to 33,897,434 bp, one QTL with pleiotropic effects for CWT, BFT, and LMA was detected (Table 1, Figures 1). On the region, one gene, BBS7 (Bardet-Biedl syndrome 7), was located. The QTL region was previously reported in other studies (McClure et al., 2010; Mizoguchi et al., 2006; Setoguchi et al., 2009). However, further studies are needed to characterize the QTL, i.e. with effects of pleiotropy of one gene or of multiple genes that are clustered together in the QTL region.
In conclusion, our results suggest that the LDVCM approach is beneficial in detection of QTL for carcass quality in a Hanwoo population, under whose conditions availability of marker density and exploitation of LDs (genetic structure) are limited. The detected QTL regions need to be verified in other Hanwoo populations as well as by conducting candidate gene studies, before implementing marker-assisted selection in Hanwoo commercial populations.
ACKNOWLEDGEMENTS
This research was supported by the Technology Development Program for Agriculture and Forestry, Ministry of Agriculture, Forestry and Fisheries, Republic of Korea, 2011. The projects was titled as “Development of production technologies for high quality and nutritional values of beef in Hanwoo” and “Development of a DNA kit for selection on growth and meat quality at early stages in Hanwoo”.