Characterization of the porcine Nanog 5′-flanking region
Article information

Abstract
Objective
Nanog, a homeodomain protein, has been investigated in humans and mice using embryonic stem cells (ESCs). Because of the limited availability of ESCs, few studies have reported the function and role of Nanog in porcine ESCs. Therefore, in this study, we investigated the location of the porcine Nanog chromosome and its basal promoter activity, which might have potential applications in development of ESCs specific marker as well as understanding its operating systems in the porcine.
Methods
To characterize the porcine Nanog promoter, the 5′-flanking region of Nanog was isolated from cells of mini-pig ears. BLAST database search showed that there are two porcine Nanog genomic loci, chromosome 1 and 5, both of which contain an exon with a start codon. Deletion mutants from the 5′-flanking region of both loci were measured using the Dual-Luciferase Reporter Assay System, and a fluorescence marker, green fluorescence protein.
Results
Promoter activity was detected in the sequences of chromosome 5, but not in those of chromosome 1. We identified the sequences from –99 to +194 that possessed promoter activity and contained transcription factor binding sites from deletion fragment analysis. Among the transcription factor binding sites, a Sp1 was found to play a crucial role in basal promoter activity, and point mutation of this site abolished its activity, confirming its role in promoter activity. Furthermore, gel shift analysis and chromatin immunoprecipitation analysis confirmed that Sp1 transcription factor binds to the Sp1 binding site in the porcine Nanog promoter. Taken together, these results show that Sp1 transcription factor is an essential element for porcine Nanog basal activity the same as in human and mouse.
Conclusion
We showed that the porcine Nanog gene is located on porcine chromosome 5 and its basal transcriptional activity is controlled by Sp1 transcription factor.
INTRODUCTION
Embryonic stem cells (ESCs), derived from inner cell mass (ICM) of a blastocyst, can proliferate indefinitely in vitro (self-renewal) and are maintained under the regulation of several key genes that are mainly expressed in pluripotent cells. These unique properties make them exceptionally valuable for cell replacement therapies, drug discovery and regenerative medicine [1]. Nanog is a homeodomain-bearing protein that acts as a transcription factor [2] and involved in pluripotent cell-specific transcription that plays a crucial role in maintaining the undifferentiated state of early postimplantation embryos and ESCs [3]. Homeobox genes have been demonstrated to be important in patterning and lineage specification during early embryogenesis [4]. Elevated levels of Nanog can maintain the self-renewal of murine ESCs, independent of leukaemia inhibitory factor (LIF), and enable growth of human ESCs without feeder cells [5].
Some exogenous growth factors support the pluripotency of ESC. For example, LIF supports the undifferentiated state of mouse ESCs by activating the signal transducer and activator 3 [6]. Another external factor known to support the self-renewal of murine ESCs is bone morphogenetic protein 4 (BMP4). In the presence of LIF, BMP4 can enhance the self-renewal and pluripotency of mouse ESCs by activating members of the Id (inhibition of differentiation) gene family [7]. However, LIF is not sufficient to maintain human ESCs under conditions that would promote self-renewal of murine ESCs [8], and BMPs cause rapid differentiation [9]. The POU domain-containing Oct4 [10] and the HMG domain-containing SOX2 are two other transcription factors known to be essential for normal pluripotent cell development and maintenance [11]. The Oct4 and Sox 2 binding sites are located within the differentially regulated Nanog promoter region and responsible for its transcriptional activity [12]. Although these transcription factors have independent roles in determining other cell types [13], at least a portion of their function in pluripotent cells occurs via a synergistic cooperation for its target genes. However, overexpression of Oct4 and Sox-2 has little effect on rescuing Nanog promoter activity in differentiated cells, indicating that some transcription factors besides Oct4 and Sox2 may be required for regulation of Nanog expression [14].
The GC-rich box to which Sp1 binds is one of the most common regulatory elements distributed in promoters of numerous housekeeping as well as tissue specific genes [15]. Sp1 can stimulate the transcription from proximal promoters and from distal enhancers [16]. It is well understood that even though ESCs of mammals (e.g., human and murine) share common stem cell markers, each species are believed to possess their own specific transcription programs [17,18] as it reported in the differential Nanog expression patterns in the ICM between humans and mice [19].
Porcine ESC lines still have not characterize, despite numerous reports of the isolation and propagation of them. Because conventional pluripotency markers are nonspecific for porcine embryonic-derived cell lines, the porcine Nanog gene might be an efficient pluripotency marker when porcine ESC lines are established. In the pig genome, two Nanog related sequences are annotated; specifically, sequences of chromosome 1 encode one exon, whereas those of chromosome 5 contain 4 exons, like other mammals. In a previous study, porcine Nanog promoter was cloned and sequenced, after which it was mapped on chromosome 5 [20], while another study mapped porcine Nanog on chromosome 1 and identified the sequences of chromosome 5 as a non-functional pseudogene [21]. Nevertheless, these studies have not addressed the promoter activity of the 5′-regulatory regions of the porcine Nanog gene. Therefore, in this study, we compared two genomic loci of tentative Nanog 5′-flanking regions, i.e., chromosome 1 and 5, in terms of promoter activity and verify its basal regulatory element. DNA sequences of chromosome 5 showed promoter activity, but those of chromosome 1 did not, and we confirmed that transcription factor Sp1 is essential to Nanog basal transcriptional activity.
MATERIALS AND METHODS
Isolation of porcine Nanog genomic DNA
For isolation of the 5′-flanking regions of the porcine Nanog gene, porcine genomic DNA was isolated from cells of miniature pig ears using the Wizard Genomic DNA purification kit (Promega, Madison, WI, USA) and used as template DNA for polymerase chain reaction (PCR) amplification. Gene-specific primers were used for PCR amplification to obtain a 1,800 bp-upstream DNA fragment containing the ATG translation start site of the porcine Nanog gene (Table 1). PCR amplification was performed in a 50 μL reaction mixture containing 1 μL of porcine genomic DNA, 10 pmol of each of the primers, and AccuPower Pfu PCR PreMix (Bioneer, Daejoen, Korea). Samples were subjected to the following PCR conditions: 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 58°C for 30 s, extension at 72°C for 2 min, and then final extension at 72°C for 5 min. Isolation and analysis of positive cloned DNA by restriction enzyme mapping was performed using the pGEM-T easy vector system (Promega, USA), followed by complete sequencing in both directions (GenBank Accession No. NC_010443, EF522119).
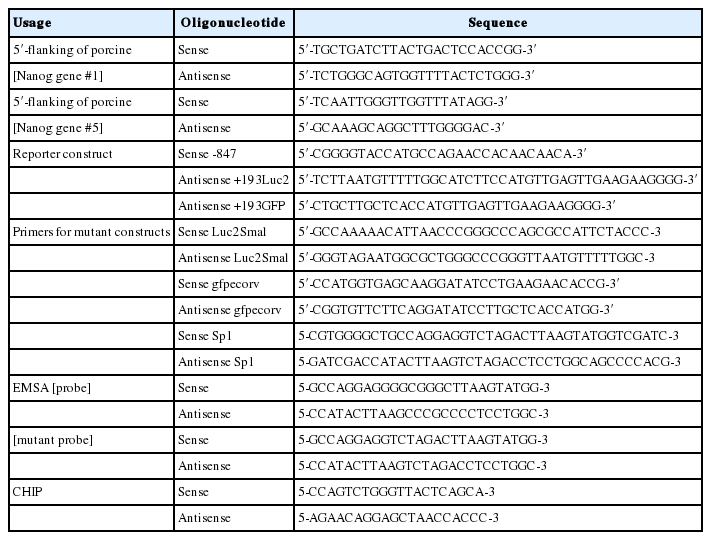
Oligonucleotides used in this study
Site-directed mutagenesis of porcine Nanog promoter sequences
Point mutations were generated using the “Quick Change” site-directed mutagenesis kit (Stratagene, Santa Clara, CA, USA) according to the manufacturer’s instructions with the primers shown in Table 1. pGL4. 10 (Promega, USA) was used as the backbone vector for luciferase activity. The vector was mutated to make Luc2_Sma I by site-directed mutagenesis in which Sma I restriction enzyme site is introduced in the reporter gene. The following is PCR conditions for site directed mutagenesis: 95°C for 30 s, followed by 18 cycles of denaturation at 95°C for 30 s, annealing at 53°C for 1 min, extension at 68°C for 5 min, and then final extension step at 68°C for 5 min. phrGFP II-C (Agilent Technologies Genomics, Santa Clarara, CA, USA) was used as the backbone vector for green fluorescence protein (GFP) expression. The vector was mutated to make phrGFPKI-PS vector by site-directed mutagenesis in which EcoRV site is introduced at the GFP open reading frame. The following is PCR conditions for site directed mutagenesis: 95°C for 30 s, followed by 16 cycles of denaturation at 95°C for 30 s, annealing at 53°C for 1 min, extension at 68°C for 5 min, and then final extension at 68°C for 5 min.
Promoter-luciferase reporter constructs and site-directed mutagenesis
The 5′-flanking region (−847/+194) of the porcine Nanog gene was cloned into the KpnI and SmaI sites of the Luc2_smaI vector and into the KpnI and EcoRV sites of the hrGFPKI-PS vector. For amplification of plasmid DNA constructs, PCR was performed using the 1,800 bp-upstream DNA fragment containing ATG as a template with primers. Briefly, PCR was performed using synthetic oligonucleotides that incorporated a 5′-end KpnI site to obtain 5′-end-deleted DNA fragments. KpnI/BlnI digestion of the fragment (−847/+194) is used to make pNANOP −644/+194 and KpnI/EcoRI digestion is for pNANOP −99/+194 deletion fragment vector. For construction of the Sp1 mutant vector, the oligonucleotide sequences were replaced by introduction of the XhoI enzyme site using a method based on site-directed mutagenesis PCR with pNANOP −99/+194 Luc2 as a template using the primers shown in Table 1. Nucleotide sequencing was performed to verify the sequences and orientations of the constructs.
Cell culture
The mouse ESC line R1 was cultured on mouse primary embryonic fibroblast feeder cells that were treated with mitomycin C (Sigma, St. Louis, MO, USA) and grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA, USA) supplemented with 15% fetal calf serum (Hyclone, Logan, UT, USA), 1% non-essential amino acids, 1% penicillin/streptomycin, 0.1 mM 2-mercaptoethanol, 1,000 U/mL LIF (ES-GRO; Chemicon, Temecula, CA, USA), 2 mM L-glutamine and 1 mM MEM sodium pyruvate solution (Gibco, USA) at 37°C under 5% CO2. Human embryonic kidney (HEK) 293T cells (ATCC: CRL 11268; Manasas, VA, USA) were maintained in DMEM (Gibco, USA) supplemented with 10% fetal calf serum (Hyclone, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco, USA) at 37°C under 5% CO2.
Transient transfection and luciferase reporter assay
HEK 293T cells were seeded at 5×104 cells per well in 24 well plates the day before transfection. Additionally, 0.5 μg each of the promoter reporter constructs was co-transfected with 0.02 μg of the Renilla luciferase vector (pRL-TK from Promega, USA) as an internal control by using jetPEI (Polyplus, New York, NY, USA). Cells were harvested after 24 h, and the luciferase activity was then measured using a dual-luciferase assay system (Promega, USA) and the LuBi Microplate Luminometer (Microdigital, Sungnam, Korea). The luciferase activity of each construct was then normalized with that of the control vector pGL4-Basic. All transfections were repeated in triplicate.
Western blot analysis
The cells were harvested and lysed using radio-immunoprecipitation assay buffer. The total protein concentration was measured with a Pierce BCA protein assay kit (Thermo Scientific, Carlsbad, CA, USA). Cell extracts (20 μg/lane) were separated in 12% sodium dodecyl sulfate polyacrylamide gel and transferred to polyvinylidene difluoride membranes with an Iblot transfer device (Invitrogen, Carlsbad, CA, USA). After blocking with 5% skim milk in Tris-buffered saline for 1.5 h at room temperature, the membranes were incubated with rabbit anti-hrGFP (1:5,000) (Agilent, USA) antibody overnight at 4°C, then with rabbit horseradish peroxidase-conjugated secondary antibody (1:2,000) (Cell Signaling Technology, Danvers, MA, USA) for 90 min at room temperature. Signals were visualized with a Super signal West Pico Chemiluminescent Substrate kit (Thermo Scientific, USA). Anti-β actin (1:1,000) antibody was used as a loading control.
Extraction of nuclear proteins
Nuclear extracts were prepared from HEK 293T cells according to the following protocols. Briefly, cells were scraped with cold phosphate buffered saline and centrifuged, then mixed with 500 μL of wash buffer (10 mM 4-[2-hydorxyethyl]piperazine-1-ethanesulfonic acid [HEPES]) pH7.9, 10 mM KCl, 2 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were then centrifuged at 3,000 rpm for 5 min at 4°C, after which the supernatant was removed. The pellet was subsequently suspended with 500 μL of buffer B (10 mM HEPES pH 7.9, 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, 0.2% NP40, 1 mM DTT, 1 mM PMSF), then incubated on ice for 5 min. Next, the pellets were centrifuged at 6,000 rpm for 5 min at 4°C, after which the supernatant was removed. The pellet was then washed twice with 500 μL wash buffer, after which the supernatant was removed. The pellet was subsequently resuspended in 80 μL of extraction buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 400 mM NaCl, 1 mM DTT, 1 mM PMSF) and vortexed the sample for 30 min in the cold room. And then it made in 20% glycerin solution and stored at −70°C.
Electrophoretic mobility shift assay
Nuclear extracts were prepared as previously described, after which the probes shown in Table 1 were annealed to their respective antisense strands. A Lightshift Chemiluminescent EMSA kit (Thermo Scientific, USA) was used for the binding reactions, in which 2 μg of nuclear extract was incubated with binding reaction buffer in the presence or absence of competitor (0.5 μg Poly(dI·dC)) for 10 min at room temperature. Biotin labeled, double-stranded oligomers were then added to the reaction mixture and incubated for an additional 20 min at room temperature. For the competition experiments, unlabeled competitors containing intact or mutant Sp1 consensus sequences were added at 200 fold excess. The reaction products were electrophoresed at 100 V in a 4% polyacrylamide gel and 0.5×tris-borate-EDTA. After transfer to a nylon membrane at 380 mA for 30 min, the membrane was cross-linked using an UV-light cross-linker, then visualized with a chemiluminescent nucleic acid detection module (Thermo Scientific, USA) according the manufacturer’s instructions.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s instructions. Briefly, nuclear lysates from porcine ear feeder cells (PEF cells) were sonicated, after which cross-linked proteins were immunoprecipitated by incubation with antibodies against Sp1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). A DNA sample from sonicated nuclear lysates was treated with proteinase K, then used as an internal control (input). A DNA sample that was immunoprecipitated with normal immunoglobulin G was used as a negative control. Immunoprecipitated DNA was detected by PCR amplification using CHIP primers (Table 1) and rTaq Plus PCR Master Mix (ELPIS BIOTECH, Daejeon, Korea). For PCR, samples were subjected to 95°C for 5 min, followed by 34 cycles of 95°C for 30 s, 62°C for 30 s, and 72°C for 30 s, and then 5 min of final extension at 72°C. The PCR products were then analyzed by 2% agarose gel electrophoresis.
RESULTS
Cloning and confirmation of porcine Nanog gene locus
Genomic sequences of chromosome 1 revealed it contain only 1 exon, while chromosome 5 was found to contain 4 exons (Figure 1A). The sequence match value between the first exon of chromosome 1 and exons from chromosome 5 containing the ATG start codon was almost same.
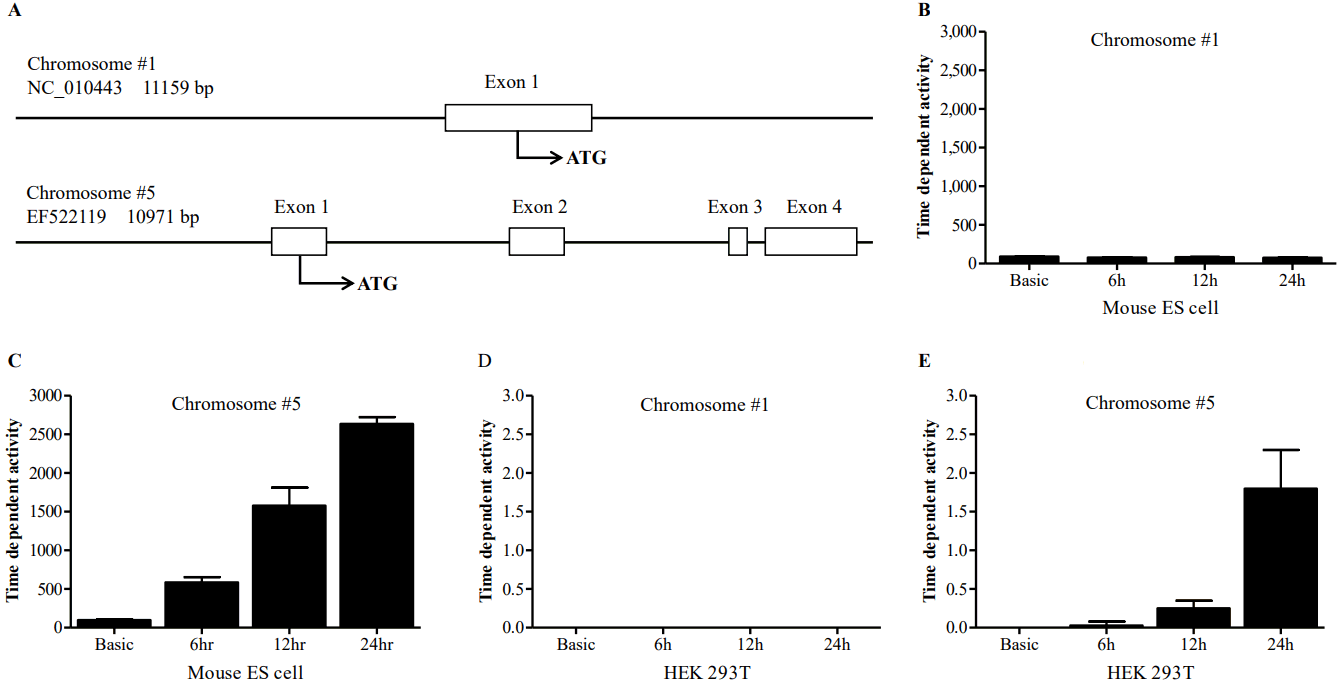
Chromosome locus and promoter activity of the porcine Nanog. (A) Confirmation of the porcine Nanog gene locus at Chromosome 1 and Chromosome 5. Both chromosome 1 and Chromosome 5 sequences possess an exon containing ATG. Sequence annotation showed that there is one exon in chromosome 1, whereas chromosome 5 has four exons. Time dependent promoter activity of the sequences of chromosome 1 (B) and those of chromosome 5 (C) in murine embryonic stem cells (ESCs), as well as in HEK293T cells (D and E, respectively) using luciferase assay.
Characterization of the porcine Nanog promoter
To find the regulatory region crucial to the transcriptional activity of the gene, promoter activities of the 5′-flanking region of chromosome 1 and chromosome 5 were measured. The 5′-flanking region of each chromosome was cloned in to the luciferase constructs (pNANOP1 Luc2 and pNANOP5 Luc2). As shown in Figure 1B to 1E, luciferase activity was only detected from pNANOP5 Luc2, and this increased in a time dependent fashion in both ES and HEK293T cells. Therefore, the DNA fragment from chromosome 5 was used for further study. The 5′-flanking region of chromosome 5 was cloned in to the GFP vector (Figure 2A) to further confirm the promoter activity (Figure 2B), and the GFP protein was detected in the cell lysates from pNANOP-847/+194 GFP transfected cells not in control cells (Figure 2C).
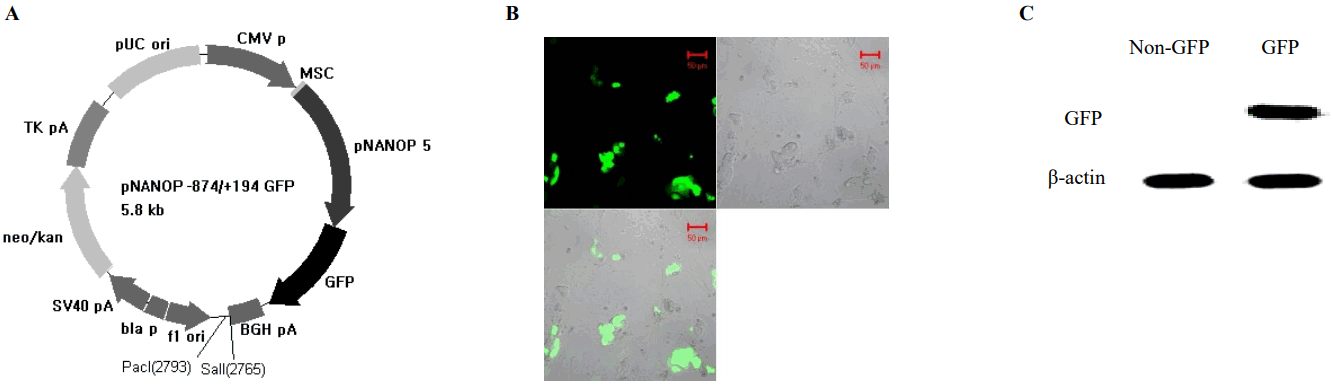
GFP expression from the construct containing the 5′-flanking region (−874/+194) from chromosome 5. (A) Vector designed (B) expression of GFP in murine embryonic stem cells (ESCs). Cells were counterstained with DAPI (1 μg/mL, Invitrogen, Carlsbad, CA, USA) and examined with appropriate filters. Images were obtained with a LSM 510 META Confocal Laser Scanning Microscope. (C) Western blot analysis. Cells were harvested, incubated with rabbit anti-hrGFP and then rabbit horseradish peroxidase-conjugated secondary antibody. Signals were visualized with chemiluminescent substrate. β-Actin was used as a loading control.
Deletion analysis of luciferase reporter gene constructs
A series of deletion constructs (Figure 3A) were generated to define the mechanisms controlling transcriptional regulation of the porcine Nanog promoter in murine ESCs and HEK293T cells. The relative luciferase expression driven by each of the constructs was normalized using a vector containing the Renilla luciferase gene as an internal standard for transfection efficiency. Approximately 10-fold higher promoter activity was observed in the wild-type construct, pNANOP-847/+194 Luc2, compared with the vector control (pGL4-basic) (p<0.05). The results from transfection experiments with murine ESCs and HEK293T cells suggest that the upstream regions in the porcine Nanog promoter, including from −847 to −644, may contain negative cis-regulatory elements, while the −99/+194 regions may contain positive cis-regulatory elements involved in regulating transcription activity of the Nanog promoter.
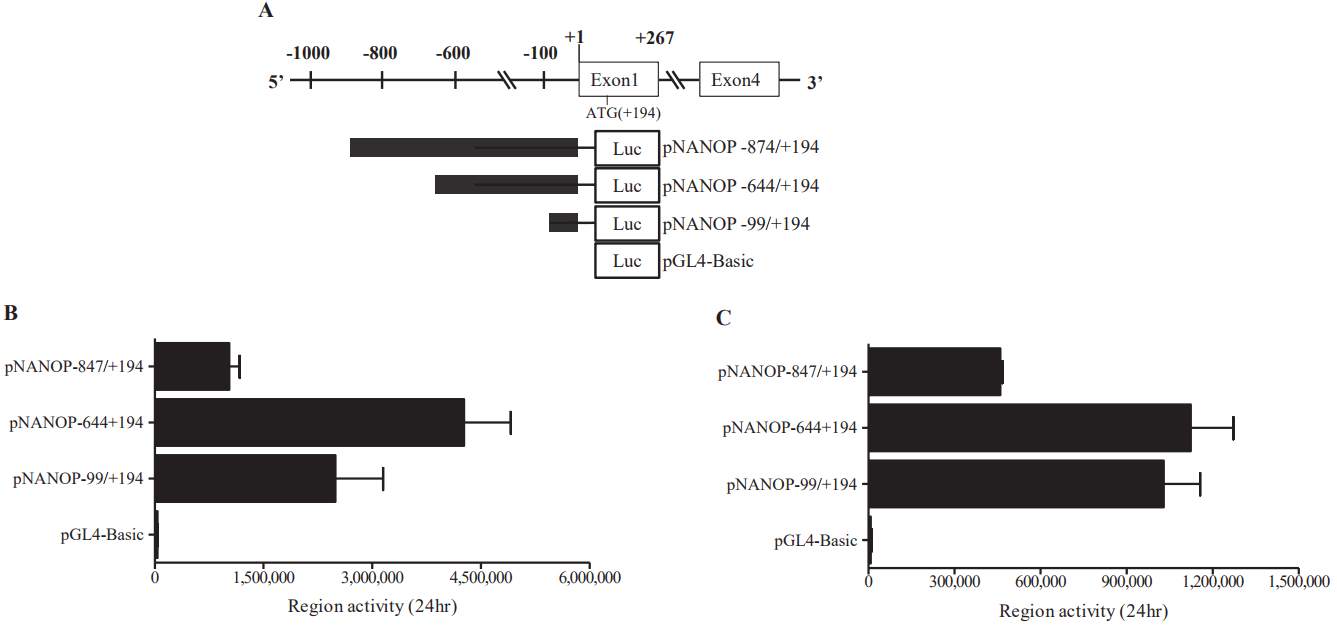
Deletion fragment of proximal promoter assay. (A) Vector designed for deletion fragments. Promoter assays with deletion fragments in murine embryonic stem cells (ESCs) (B) and in HEK293T cells (C).
Sp1 is essential in modulating porcine Nanog gene transcription
Sequences of promoter region were analyzed using TRANSFAC for the DNA motifs recognized by the known transcription factors. Sequences were aligned with human, mouse, bovine, and porcine Nanog 5′-proximal promoter regions. Comparison of the sequences of porcine and other mammalian Nanog promoters (e.g., human, murine, and bovine) revealed another putative transcription factor binding site between Oct4/Sox2 and the transcription start site; specifically, one NF-κB transcription factor binding site and one Sp1 binding site between the NF-κB binding site and the transcription start site (Figure 4A). To examine the role of the Sp1-binding site, we performed mutagenesis to introduce mutations of Sp1 (GGCGGG → TC TAGA) binding sites generating the pNANOPSPm −99/+194 construct (Figure 4B). Ablation of the Sp1 binding site resulted in an approximately 80% reduction in Nanog promoter activity (p<0.05) in ESCs (Figure 4C), as well as in HEK293T cells (Figure 4D). These findings suggest that the Sp1 binding site plays a role in porcine Nanog transcriptional activation.
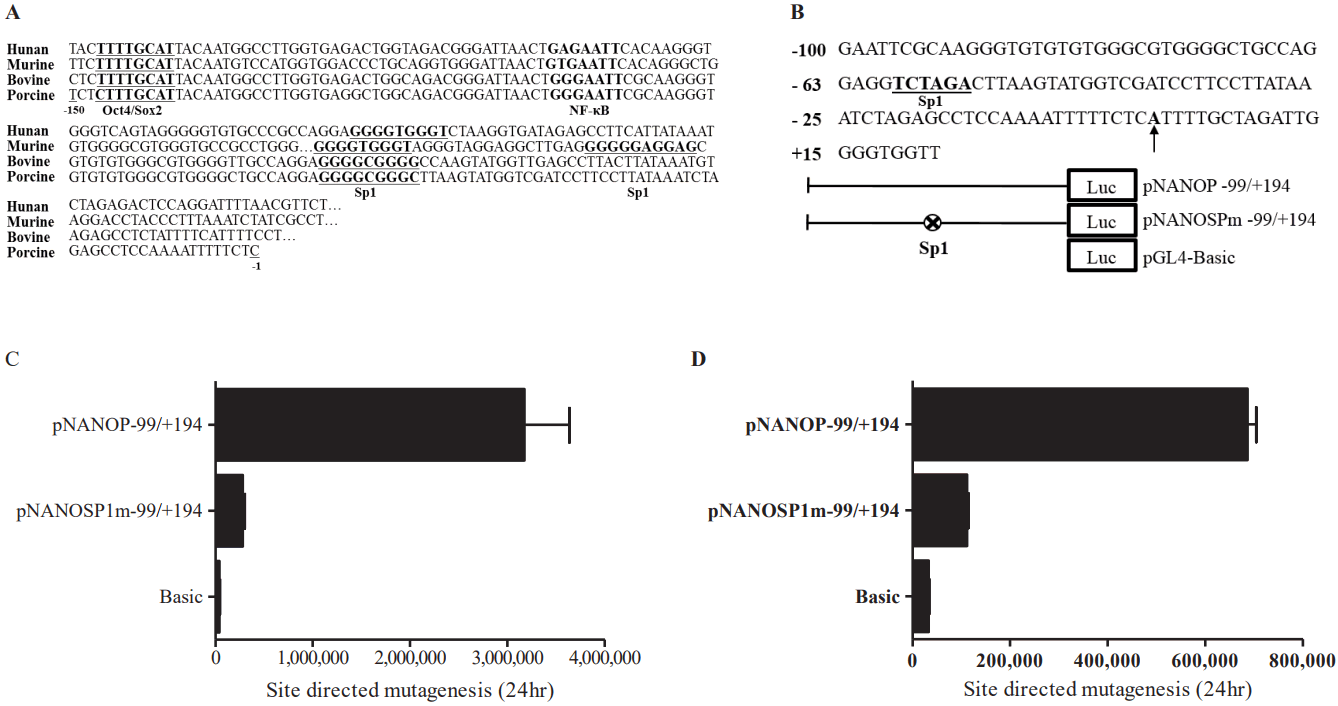
Sequence analysis and site-directed mutagenesis assay. (A) Comparative analysis of Nanog 5′-proximal promoter sequences. Oct4/Sox2, underline; NF-κB, bold; Sp1, bold/underline. (B) Construction of the Sp1 mutant constructs, the oligonucleotide sequences were replaced by introduction of the XhoI enzyme site. pNANOP-99/+194 and pNANOSPm-99/+194 constructs and pRL-TK were transfected into embryonic stem cells (ESCs) (C) and HEK 293T cells (D). Cells were harvested after 24 h, after which the luciferase activity was measured (* p<0.05).
Direct binding of Sp1 to the Sp1-binding site in the porcine Nanog promoter
To evaluate the ability of Sp1 to recognize the Sp1 binding site within the porcine Nanog promoter, electrophoretic mobility shift assay (EMSA) was performed using a 20 bp probe spanning the Sp1 binding site combined with nuclear extracts from PEF cells. Use of this probe resulted in observation of a protein-DNA complex, which was found to compete in the presence of a 100-fold excess of the unlabeled wild-type probe (Figure 5A). In addition, mutated Sp1-mut probes were used as competitors. The shifted bands were eliminated when incubated with 100-fold excess unlabeled probe, but the mutated-unlabeled probe had no effect, indicating the specificity of the Sp1 binding site (Figure 5A). Sp1 specific binding was confirmed by the decreased intensity of the Sp1 bands. To further validate the binding between Sp1 in vivo, we conducted ChIP assay using PEFs (Figure 5B). Taken together, these data suggest that the activity of the putative basal porcine Nanog promoter is regulated by SP1 transcription-factors in vivo.

Gel shift and chromatin immunoprecipitation (ChIP) analyses of nuclear proteins binding to the Sp1 binding sites. (A) Electrophoretic mobility shift assay (EMSA). Lane 1, probe Sp1 incubated without embryonic stem cells (ESCs) nuclear extract proteins; Lane 2, probe Sp1 incubated with ESCs nuclear extract proteins; Lane 3 and 4, specific and nonspecific competition. (B) Chromatin immunoprecipitation assay demonstrating the in vivo potential of Sp1 to bind to Nanog and Sp1 elements, respectively.
DISCUSSION
Nanog has been reported to be a key transcription factor responsible for maintaining pluripotency [2]. Nanog is transcribed specifically in pluripotent cells in mouse pre-implantation embryos, ESCs, and embryonic germ cells [22], as well as monkey and human ESCs [23]. Some E in bovine blastocysts express Nanog protein [24]. Nevertheless, we still do not know well for the Nanog expression and its function in porcine despite numerous reports of the isolation and propagation of putative pESC lines.
In this study, we analyzed the 5′-flanking region of the porcine Nanog gene to enable better understanding of the transcriptional regulation of the porcine Nanog gene. First, we sought to confirm the porcine Nanog genomic locus using BLAST database, as both porcine chromosome 1 and 5 are predicted to contain the Nanog gene. These two regions have the first exon containing ATG, even though chromosome1 has one exon and chromosome 5 has four exons. Using mouse ESCs and HEK293T as heterologous systems, we found that the genomic sequences from chromosome 5 had promoter activity, while those from chromosome 1 did not. This result is in agreement with a previous study [20] but not with a recent one [21]. To clarify the discrepancy of these results, further investigation warrants.
The Oct-4 and Sox-2 binding sites are located within the differentially regulated Nanog promoter region and responsible for its activity [12]. A reporter gene driven by the Nanog proximal promoter containing the Oct4/Sox2 motif recapitulates Nanog transcription trends in both pluripotent and non-pluripotent cells, confirming that this motif is well-preserved between mice, rats and humans. Analysis of the 5′-flanking region of the porcine Nanog promoter revealed that the Oct4/Sox2 motif was localized at −150 bp upstream of the transcription start site. Additionally, luciferase activity of pNANOP- 644/+194 with the Oct4/Sox2 motif was higher than that of pNANO-99/+194. These findings suggest that Oct4/Sox can be act to promote Nanog transcription in porcine as well.
Sp1 is a member of the Sp1-like/krüppel like factor family, which plays an important role in expressed transcription factors. Members of this family are implicated in the regulation of diverse cellular functions [25] and participate in the activation of Oct-4 expression [14]. We found that the regulatory sequences of the porcine Nanog gene contain the Sp1 binding site at -61. The role of the Sp1 binding site was confirmed through both deletion and mutagenesis analyses. Binding of Sp1 transcription factor was further confirmed by gel shift and ChIP analyses. Based on these findings, we have confirmed the transcriptional regulation of porcine Nanog 5′-proximal region and its location on chromosome that might be useful observations to develop a maker for preimplantation embryonic development as well as ESCs establishment in pig.
CONCLUSION
In this study, we examined the promoter activity of tentative porcine Nanog regulatory sequences found in both chromosome 1 and 5, and found that sequences from chromosome 5 possess promoter activity, while those from chromosome 1 do not. In addition, we identified the Sp1 transcription factor binding sites that are located in the 5′-regulatory sequences of the porcine Nanog promoter by EMSA, ChIP and deletion mutant analyses, confirming that Sp1 plays a crucial role in basal transcriptional activity of the porcine Nanog promoter.
ACKNOWLEDGMENTS
This research was supported by grants from the Next-Generation 21 Program (Project No. PJ01109301), Rural Development Administration, Republic of Korea.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.