Dietary Niacin Supplementation Suppressed Hepatic Lipid Accumulation in Rabbits
Article information
Abstract
An experiment was conducted to investigate the effect of niacin supplementation on hepatic lipid metabolism in rabbits. Rex Rabbits (90 d, n = 32) were allocated to two equal treatment groups: Fed basal diet (control) or fed basal diet with additional 200 mg/kg niacin supplementation (niacin). The results show that niacin significantly increased the levels of plasma adiponectin, hepatic apoprotein B and hepatic leptin receptors mRNA (p<0.05), but significantly decreased the hepatic fatty acid synthase activity and adiponectin receptor 2, insulin receptor and acetyl-CoA carboxylase mRNA levels (p<0.05). Plasma insulin had a decreasing tendency in the niacin treatment group compared with control (p = 0.067). Plasma very low density lipoproteins, leptin levels and the hepatic adiponectin receptor 1 and carnitine palmitoyl transferase 1 genes expression were not significantly altered with niacin addition to the diet (p>0.05). However, niacin treatment significantly inhibited the hepatocytes lipid accumulation compared with the control group (p<0.05). In conclusion, niacin treatment can decrease hepatic fatty acids synthesis, but does not alter fatty acids oxidation and triacylglycerol export. And this whole process attenuates lipid accumulation in liver. Besides, the hormones of insulin, leptin and adiponectin are associated with the regulation of niacin in hepatic lipid metabolism in rabbits.
INTRODUCTION
Liver plays a key role in lipid metabolism and is the hub of fatty acid synthesis and lipid circulation through lipoprotein synthesis in mammals. Triacylglycerols may accumulate in hepatocytes or are exported as constituents of very low density lipoproteins (VLDL) (Nguyen et al., 2008). The excessive accumulation of lipid droplets into the hepatocytes can result in hepatic steatosis. The triacylglycerol content of hepatocytes is regulated by the activity of cellular molecules that facilitates hepatic fatty acid synthesis and esterification (‘input’), and hepatic fatty acid oxidation and triacylglycerol export (‘output’). The genes encoding levels of Acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS) and carnitine palmitoyltransferase (CPT) 1 were respectively involved in the regulation of FAS and oxidation. Besides, the lipid metabolism in liver in mammals can be regulated by hormones (e.g., insulin, leptin, and adiponectin) (Saltiel and Kahn, 2001; Berg et al., 2002; Gallardo et al., 2007; Peng et al., 2009) and nutritional state (carbohydrates, B vitamins) (Havel, 2004; Ganji et al., 2015).
Emerging findings indicate that niacin decreases plasma VLDL level by inhibiting hepatocyte diacylglycerol acyltransferase 2 (Kamanna et al., 2013). Lamon-Fava et al. (2008) found that niacin treatment significantly increased high density lipoprotein (HDL) apo-protein A-I (ApoA-I) concentrations and production, as well as enhanced the clearance of triglyceride-rich lipoproteins. G-protein-coupled receptor 109 A (GPR109A) was identified as receptors for niacin (Wise et al., 2003). Hepatic-specific over-expression of GPR109A in mice reduced plasma HDL cholesterol accompanied by reduction in hepatic cholesterol efflux to ApoA-I (Li et al., 2010). Generally, the studies of niacin were focused on relative lipid regulation in plasma for the treatment of dyslipidemia and atherosclerosis. But the effect of niacin on hepatic fatty acid metabolism is still unclear.
Obesity and obesity-associated fatty liver disease are becoming global health problems in adults as well as children (Browning et al., 2004; Hamaguchi et al., 2005). Hepatic steatosis is encountered in about 20% to 35% of the general adult population in the United States (Reddy and Rao, 2006). The prevention or treatment of hepatic steatosis is difficult to implement, especially by nutrition intervention methods. In the present study, we investigated the effect of dietary niacin addition on hepatic fatty acid metabolism in rabbits, and determined the possibility of niacin attenuates hepatic lipid accumulation.
MATERIALS AND METHODS
Animals
Three-month-old Rex-rabbits were individually housed in locally made cages (60×40×40 cm). Temperature and lighting were maintained according to commercial conditions. The diets were formulated according to the values for growing rabbits from NRC (1977) and de Blas and Mateos (1998). The diets were pressure pelleted with the diameter of the pellets being 4 mm. All rabbits received a starter diet containing 16% crude protein, 14% crude fiber and 11 MJ/kg of digestible energy. All rabbits had free access to food and water during the rearing period. This study was approved by the Shandong Agricultural University and conducted in accordance with the “Guidelines for Experimental Animals” of the Ministry of Science and Technology (Beijing, China).
Experimental protocol and sample collection
At 90 days of age, 32 rabbits of similar body weight (1,860±100 g) were divided into 2 groups (16 replicates per group and 1 rabbit per replicate): Fed basal diet (control, measured niacin content was 28 mg/kg) or fed basal diet with 200 mg/kg niacin addition (niacin, measured niacin content was 230 mg/kg). The experiment lasted for 8 wks which included 1 wk adaptation period and 7 wks experimental period. At the end of the trial, 16 rabbits (8 rabbits per group, 4 male and 4 female, with the average body weight of the 8 rabbits equal to the average body weight of entire treatment group) were electrically stunned (70 V, pulsed direct current, 50 Hz for 5 s), and 10 mL blood was collected immediately from the heart. Plasma was obtained following centrifugation at 400 g for 10 min at 4°C and stored at −20°C for subsequent analysis. The rabbits were sacrificed by cervical dislocation, and the liver was collected. After being snap-frozen in liquid nitrogen, the liver tissue samples were stored at −80°C.
Measurements
Plasma VLDL concentration was determined using the method described by Barter and Lally (1978). Plasma adiponectin and leptin concentrations were determined using a validated sandwich enzyme-linked immunosorbent assay (Uscn Life Science Inc., Wuhan, China) with an adiponectin- or leptin-specific antibody. Intra-assay coefficient of variation of adiponectin was 2.3%, and leptin was 3.0%.
Plasma insulin was measured by radioimmunoassay with a guinea pig anti-porcine insulin serum (3 V Bioengineering group Co., Weifang, China). In this procedure, 125I-labelled porcine insulin competes with chicken insulin for sites on anti-porcine insulin antibodies that are immobilised on the wall of a polypropylene tube. Significant cross-reactivity has been observed between chicken insulin and guinea pig anti-porcine sera (Simon et al., 1974). The insulin in this study was referred to as immunoreactive insulin. The sensitivity of the assay was 1 μIU/mL, and all samples were included in the same assay to avoid inter-assay variability. The intra-assay coefficient of variation was 2.01%.
Liver samples were ground with saline (saline volume/sample weight = 4:1) to measure apo-protein B (Apo B) concentration and FAS activity. Apo B concentrations in liver were measured using immunoturbidimetric assays. The FAS activity in liver was measured using ultraviolet chromatometry. All the commercial diagnostic kits were from Jiancheng Bioengineering Institute (Nanjing, China).
The accumulation of cytoplasmic lipid droplets was visualised by Oil Red O staining according to the protocol of Lillie and Fullmer (1976). Briefly, tissues were immediately frozen in liquid nitrogen and cut with a Leica CM-1850 cryostat microtome (Leica, Wetzlar, Germany). Then 16-mm thick sections were fixed in 4% formaldehyde for 10 min and stained with filtered 0.5% Oil Red O (Sigma-Aldrich, St Louis, MO, USA), which was made by dissolving in isopropyl alcohol for 15 min at room temperature. Morphometric analysis was performed on 10 randomly chosen fields containing transverse sections of liver from each rabbit. The selected fields were photographed using an Olympus CX41 phase contrast microscope (Olympus, Tokyo, Japan). The volume density of each Oil Red O positive liver tissue was determined using the point-counting method described by Weibel and Bolender (1973).
Total RNA extraction and quantitative real-time polymerase chain reaction (PCR) were performed as described previously (Li et al., 2014). Sequences of primers are shown in Table 1. The PCR data were analyzed with the 2−ΔΔCT method (Livak and Schmittgen, 2001). The mRNA levels of target genes were normalised to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA (ΔCT). On the basis of the Ct values, GAPDH mRNA expression was stable across the treatments in this study (p>0.1).

Gene-specific primers used for the analysis of rabbit gene expression
Statistical analysis
The data are presented as the means±standard error of the mean, with n = 8 for plasma metabolites, hormones, hepatic FAS activity, Oil Red O staining and all mRNA levels analysis. All of the data collected were subjected to one-way analysis of variance analysis with the Statistical Analysis Systems statistical software package (Version 8e, SAS Institute, Cary, NC, USA), and the primary effect of niacin treatment was evaluated. Homogeneity of variances among groups was confirmed using Bartlett’s test (SAS Institute, USA). When the primary effect of treatment was significant, differences between means were assessed by unpaired T-Test. p<0.05 was statistically significant, and 0.05<p<0.1 was changed tendency.
RESULTS
The relative hormones and VLDL concentration in plasma were determined, and the results are shown in Figure 1. Although no significant difference was observed in plasma levels of VLDL (Figure 1A) and leptin (Figure 1B) in niacin group rabbits compared with the control, plasma adiponectin was significantly increased with 200 mg/kg niacin addition (p<0.05, Figure 1C). In addition, plasma insulin had a decreasing tendency in the niacin treatment group compared with the control group (p = 0.067, Figure 1D).
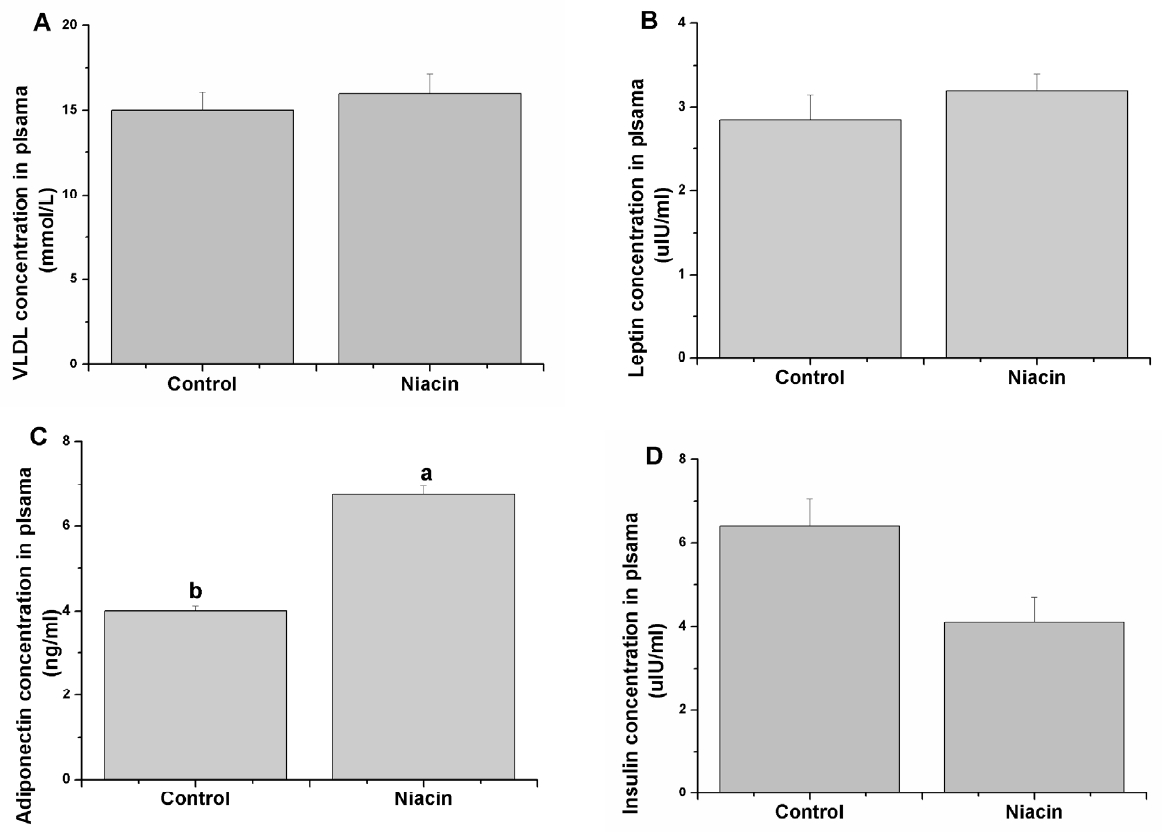
Effects of dietary niacin on plasma very low density lipoproteins (VLDL) (A), leptin (B), adiponectin (C), and insulin (D) concentration. Values are shown as the mean±standard error of the mean. a,b Means with different superscripts are significantly different (p<0.05).
Though determining the relative genes expression or enzyme activity-related lipid metabolism, we found that dietary addition of niacin significantly decreased the FAS activity (Figure 2A) and adiponectin receptor 2 (adpn R2) (Figure 2B), insulin receptor (INSR) (Figure 2C) and ACC (Figure 2D) mRNA levels (p<0.05) but increased the Apo B (Figure 2E) content and leptin receptor (LEPR) gene expression (Figure 2F) (p<0.05). Besides, the adpn R1 (Figure 2G) and CPT1 (Figure 2H) genes expression were not significantly altered when niacin addition in diet.
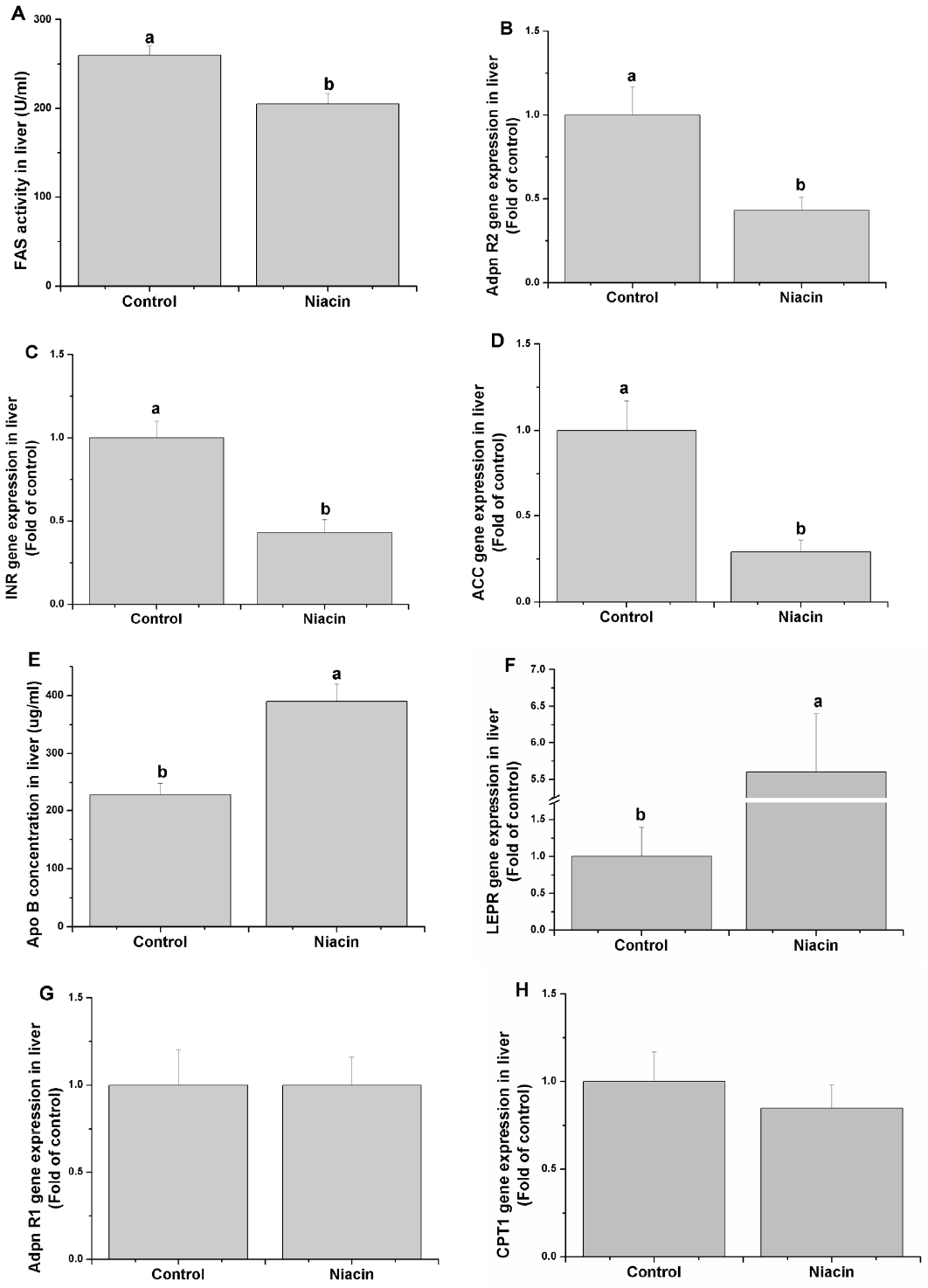
Effects of dietary niacin on hepatic fatty acid synthase (FAS) activity (A), adiponectin receptor 2 (Adpn R2) gene expression (B) insulin receptor (INSR) gene expression (C), acetyl-CoA carboxylase (ACC) gene expression (D), Apo B concentration (E), leptin receptor (LEPR) gene expression (F), adiponectin receptor 1 (Adpn R1) gene expression (G) and carnitine palmitoyltransferase (CPT) 1 gene expression (H). Values are shown as the mean±standard error of the mean. a,b Means with different superscripts are significantly different (p<0.05).
From Figure 3, we can determine that dietary niacin treatment had a significant inhibition on cytoplasmic lipid accumulation compared with control.
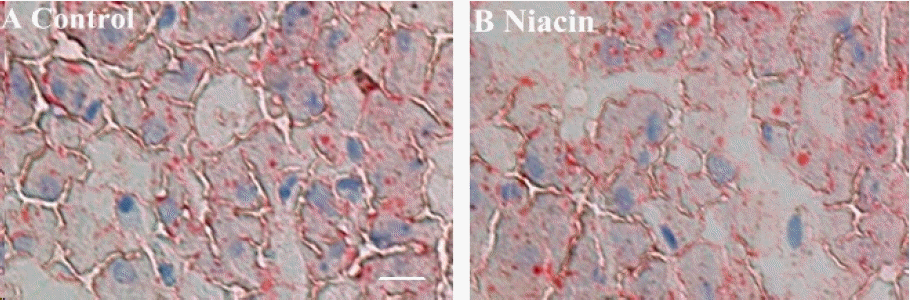
Oil Red O staining of liver for cytoplasmic lipid droplets (indicated by arrows) showing effect of dietary niacin treatment on lipid accumulation in hepatocyte (A: control, B: niacin) from rabbit. The scale bar in (A) represents 50 mm.
DISCUSSION
Dietary niacin can regulate lipid metabolism in kidney, adipose tissue and lipid transportation in plasma (Rubic et al., 2004; Cho et al., 2010; Fabbrini et al., 2010). Recent researches show that niacin increases HDL by reducing hepatic expression cholesteryl transfer protein and reducing the uptake of HDL (Jin et al., 1997; van der Hoorn et al., 2008). However, the effect of niacin on fatty acids metabolism in liver is still unclear. Our study shows that dietary niacin supplementation inhibited hepatic fatty acids synthesis and lipid accumulation.
Lipid metabolism in liver is key for energy homeostasis in rabbits. Fatty acids synthesis in liver is tightly controlled by nutritional conditions (Asai and Miyazawa, 2001). In our study, the ACC gene and FAS activity were significantly decreased in liver after niacin supplementation, which suggest that the hepatic fatty acids synthesis process is significantly inhibited by niacin. The results are consistent with the study in rats, as niacin administration relieved the increased ACC and FAS protein due to chronic renal failure in kidney (Cho et al., 2010). But the niacin treatment didn’t change the hepatic CPT1 gene expression in normal rabbit liver or human primary hepatocytes (Carling et al., 2008), which is the rate-limiting enzyme of fatty acid catabolic pathway. The results suggest that the regulation of niacin on fatty acid metabolism is only on the anabolic pathway, but not on the catabolic pathway.
Hepatic lipid components that are synthesized in smooth endoplasmic reticulum, are glycosylated in golgi apparatus, and released into blood in the form of VLDL. Apo B is the key component whose rate of synthesis in the rough endoplasmic reticulum controls the overall rate of VLDL production (Cruz-Bautista et al., 2015). Results from liver slice studies has suggested that species with limited hepatic lipogenesis have less ability to secrete VLDL from the liver compared with species in which the liver is a major or moderate source of lipogenesis (Pullen et al., 1990). But the plasma VLDL concentration was not altered after niacin administration in our experiment. The major reason might be related to the increased hepatic Apo B level. Although there was less lipogenesis, niacin can stimulate the other synthetic component (Apo B) production. Which can result in a unaltered VLDL level.
Hepatic fatty acids metabolism is also affected by hormones (e.g., insulin, leptin, and adiponectin). Many studies have found the transcription rate of the FAS gene is quickly increased after insulin administration in diabetic mouse and rats (Paulauskis and Sul, 1988; Ruzzin et al., 2010). Besides, insulin can also inhibit Apo B secretion in isolated human hepatocytes (Salhanick et al., 1991). These effects are achieved by the binding of insulin to the INSR at the cell surface (Nakae and Accili, 1999). In the present experiment, the decreased tendency of plasma insulin and significant inhibition of hepatic INSR gene in niacin group suggest that the insulin signal is associated with the regulation of niacin on hepatic lipid metabolism. But these results are inconsistent with a previous observation, which found that niacin had no effect on insulin action in human (Fabbrini et al., 2010). Perhaps the effect of niacin on insulin can be species-dependent.
Leptin is another hormone that may be involved in lipogenesis. There is a growing consensus that leptin limits fat storage not only by inhibiting food intake but also by affecting specific metabolic pathways in liver and adipose (Anania, 2002; Kitamura et al., 2006). The previous studies show that leptin can stimulate fatty acid oxidation and inhibit lipogenesis by down-regulating the expression of genes involved in fatty acid and triglyceride synthesis in liver or adipose (Bai et al., 1996; Matsusue et al., 2003; Buettner et al., 2008). Although the plasma leptin concentration was not affected by the niacin, the hepatic LEPR was increased 5-fold than control. The result shows that the hepatic leptin signal is involved to the niacin regulation on lipid metabolism.
Adiponectin, a protein exclusively secreted from adipose tissue, and its agonists could be promising candidates for the treatment of obesity-associated metabolic syndromes (Berg et al., 2002). Recent studies suggest that adiponectin can act directly on hepatic lipid metabolism (Li et al., 2014; Lin et al., 2015). Adiponectin can regulate hepatic fatty acid metabolism in vivo by attenuating the activities of FAS and ACC (Xu et al., 2003). It can also improve dyslipidemia and decrease lipid accumulation in muscle, thus enhancing insulin sensitivity in this tissue (Yamauchi et al., 2001). In present study, niacin significantly increased the plasma adiponectin level, which counters the hepatic ACC gene expression and FAS activity. It is therefore hypothesized that niacin affects lipogenesis by affecting the adiponectin secretion. Adpn R2, not Adpn R1, may be the crosslink between adiponectin signaling and nonalcoholic fatty liver disease in obese mice (Peng et al., 2009). In present study, hepatic Adpn R1 and Adpn R2 mRNA levels did not show the same tendency with plasma adiponectin level after niacin treatment. Therefore, the relationship of Adipo R with adiponectin regulating hepatic lipogenesis in niacin treated-rabbits remains unclear and warrants further investigation.
In conclusion, niacin treatment can decrease hepatic fatty acids synthesis, but not alter fatty acids oxidation and triacylglycerol export, which attenuates lipid accumulation in liver. Besides, the hormones of insulin, leptin and adiponectin are associated with the regulation of niacin on the hepatic lipid metabolism in rabbits.
ACKNOWLEDGMENTS
This work was supported by the Modern Agro-industry Technology Research system (CARS-44-B-1), Agricultural Major Application Technology Innovation Project of Shandong Province (2015–2016), Project Funded by China Postdoctoral Science Foundation (2015M580601) and Youth Science and Technology Innovation Fund of Shandong Agricultural University (2015–2016).
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.